
 is a group of rare primary immunodeficiency disorders characterized by normal or increased IgM...){kind=link}

Hyper-IgM syndrome (HIGM) is a group of rare primary immunodeficiency disorders characterized by normal or increased IgM with markedly reduced or absent IgG, IgA, and often IgE due to defective class-switch recombination (CSR) or its signalling. This disorder is caused when our immune system fails to switch from IgM to other classes of antibodies, like IgG, IgA, and IgE.
Globally, X-linked HIGM is estimated to be about 1–2 affected newborn boys per million (often cited as ~2 per million newborn boys). Although uncommon, it can lead to life-threatening infections and complications if not diagnosed and treated early.
Physiology of Antibody Production
An antibody, also called an immunoglobulin (Ig), is a protein that is produced by B-cells or plasma cells of our immune system. These antibodies “tag” the foreign particles, whether these are germs or parasites, making it easier for other immune cells to recognize them and destroy them. These antibodies are:
- IgM: the first antibody made in a new infection.
- IgA: found in mucosal secretions (saliva, gut).
- IgE: rises in allergies and parasitic infections.
- IgG: gives long-term protection and crosses the placenta.

Image licensed from Shutterstock
Normally, B-cells first produce IgM during an initial immune response. However, once activated, B-cells can “switch” to making IgG, IgA, or IgE through a process called class switch recombination (CSR).
Activation of B-Cells:
Every B-cell has thousands of receptors on its surface. Each receptor is very specific for only one type of antigen, which can be either a fragment of a virus protein or a bacterial sugar.
- B-cell activation requires co-stimulation from helper T-cells (CD4+ T-cells), another immune cell. When these cells recognize the same antigen, they express it as CD40 ligands (CD40L or CD154) on their cell surface, which bind to the CD40 receptor on B-cells to stimulate the B-cells to produce antibodies. This CD40L-CD40 interaction is also important to activate macrophages and other cellular immune responses.
- Helper T-cells also produce cytokines, the soluble messenger proteins like interleukin 4 (IL-4), IL-21, and TGF-beta, depending on the immune context. These cytokines signal the intracellular pathways to promote germline transcription at selected switch regions of DNA.

Normal humoral immune response involving IL-1, IL-4, and IL-5 signaling; defective class switching at the B-cell stage leads to Hyper IgM Syndrome.
Class Switch Recombination (CSR):
Upon activation, the DNA of B-cells undergoes rearrangements to change the class of antibody produced. This process of DNA modification for the production of specific antibodies is called class switch recombination.
Instructions for each antibody are present at different places in the B-cell’s DNA. B-cell activation stimulates the activation of the enzymes Activation-Induced Cytidine Deaminase (AID) and Uracil-DNA Glycosylase (UNG), both of which work together to cut and rejoin DNA fragments at specific points to produce a specific antibody.
Any mutation or defect in this pathway disrupts CSR, leading to continued IgM production while IgG, IgA, and IgE remain deficient. Any mutation or defect in this pathway disrupts CSR, leading to continued IgM production while IgG, IgA, and IgE remain deficient.
Causes & Pathophysiology of Hyper-IgM Syndrome
Hyper IgM syndrome results from genetic or acquired defects that prevent class switching from IgM to other antibody types.
Primary (Genetic) Causes of HIGM:
The genetic mutation causes problems either in the CD40L-CD40 interaction, in cytokine signaling, or in the enzymes required for CSR.
- X-Linked HIGM: The most common form (around 60–70% of all HIGM cases) is caused by mutations in the CD40L (CD154) gene on the X chromosome.
The mutation prevents proper CD40L expression on T-cells, disrupting CD40L–CD40 signaling needed for class switching and macrophage activation. - AID (AICDA) Deficiency: It is an autosomal recessive HIGM, in which the B-cells fail to rearrange the required DNA fragment to switch from IgM antibody to other isotopes. However, the normal CD40L-CD40 interaction doesn’t interfere with the activation of macrophages and other cellular immune responses.
- UNG Deficiency: A faulty or absent UNG enzyme prevents the normal DNA breaks for the CSR. B-cells get stuck making only IgM, resulting in hyper-IgM syndrome.
Secondary (Acquired) Causes of HIGM:
Not all the causes of hyper IgM syndrome are genetic; there are some specific diseases, treatments, and conditions that disturb the normal levels of different immunoglobulins, resulting in HIGM-like symptoms.
- HIV infection: It destroys the CD4+ helper T-cells, preventing CD40L-CD40 interaction and cytokine signaling for the CSR, resulting in the production of only IgM.
- B-cell Tumors: Tumors cause abnormal and dysfunctional B-cells, which fail to class-switch. Studies observed the decreased IgG and IgA antibodies in chronic lymphocytic leukemia (CLL).
- Therapeutic Causes: Some drugs, like rituximab,, deplete the CD20+ B-cells and their precursors. Over time, when you repeatedly use the drugs for a longer time, these decrease the IgG and IgA in the serum. However, IgM takes time to get depleted, showing the picture of relatively higher IgM levels in the body.
- Other Immunosuppressive drugs: Drugs like tacrolimus, cyclosporine, corticosteroids, and antimetabolites, indirectly disturb the antibody profile, leading to HIGM-like picture.
Symptoms of Hyper IgM Syndrome
Symptoms in most children with X-linked HIGM appear in infancy. Usually, children with HIGM present a typical pattern of severe recurrent infections early in childhood and failure to mount a normal healthy response to vaccines.
Recurrent Infections:
As the immune system is compromised, the child remains ill with different infections.
- Respiratory Disease: Recurrent upper and lower respiratory tract infections are common in HIGM patients. Children suffer repeatedly from sinusitis, middle ear infections, and bacterial pneumonia.
- Opportunistic & Unusual Infections: Because of weakened immunity, the patient becomes vulnerable to opportunistic infections, which are caused only when the individual has compromised immunity. Pneumocystis jirovecii (formerly jirovicii) pneumonia (PCP/PJP) and chronic Cryptosporidium infection are hallmark features.
Gastrointestinal (GIT) Problems:
Children and adults with HIGM syndrome often suffer from GIT problems like chronic diarrhea, usually caused by Cryptosporidium species or bacteria such as Giardia and Salmonella. This diarrhea keeps repeating, leading to malnutrition, poor weight gain, and failure to thrive in children.
Over time, malabsorption may lead to vitamin deficiencies, weakened bones, and anemia, a condition of lower-than-normal blood.
Liver & Biliary Problems:
The chronic infection of Cryptosporidium parvum often affects the liver and biliary system, which is one of the life-threatening complications of hyper-IgM syndrome. The infection can travel up into the bile ducts, leading to sclerosing cholangitis.
You may develop jaundice, abdominal pain, and itching due to bile blockage caused by sclerosing cholangitis. Over time, this can cause liver cirrhosis, in which your liver becomes hard and scarred, ultimately resulting in liver failure.
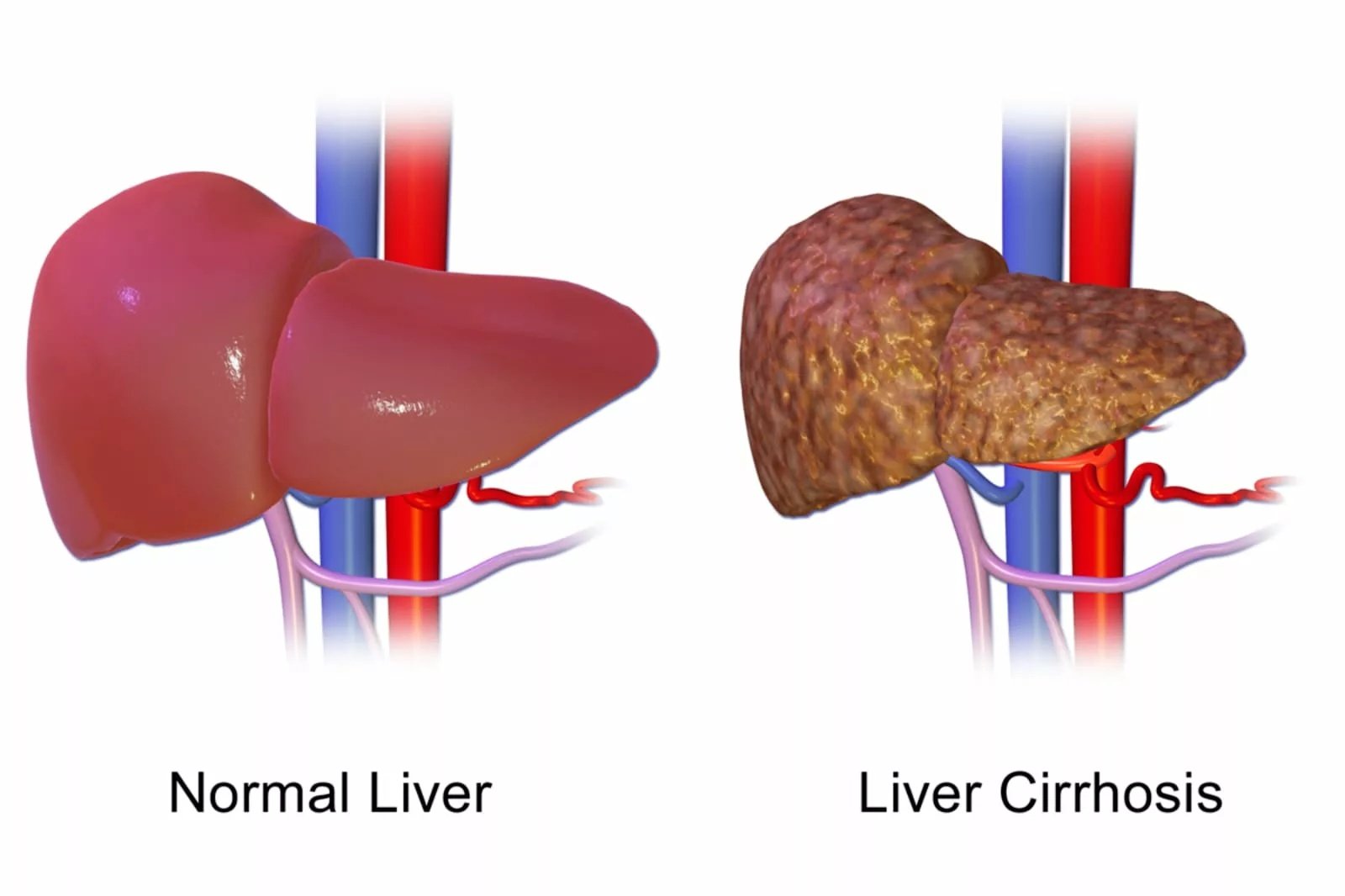
Image Credit: BruceBlaus. Liver Cirrhosis Diagram. Licensed under CC BY-SA 4.0. Source: Wikimedia Commons.
Blood & Lymphatic Symptoms:
X-linked HIGM also affects the blood and lymphatic system by causing neutropenia, a condition of low levels of neutrophils, making the patient vulnerable to different infections. Neutropenia causes mouth ulcers, gum disease, and recurrent skin infections.
These repeated infections also influence the lymphatic system, resulting in swollen lymph nodes, enlarged liver and and spleen. The impaired immune regulation may cause the patient to develop autoimmune complications in which the patient’s own immune system destroys the RBCs and platelets, leading to autoimmune hemolytic anemia or immune thrombocytopenia.
Diagnosis of Hyper IgM Syndrome
Diagnosis of the HIGM starts with suspicion of this disease when the child or infant has repeated, multiple, and opportunistic infections with poor response to routine vaccines. PCP, chronic diarrhea, especially linked with cryptosporidium, are the classical red flags.
Doctors may take the medical and family history of the patient, as HIGM can also be caused by some secondary conditions and drugs.
Initial Blood Tests:
After the suspicion, your doctor may ask for a complete blood count and quantitative immunoglobulin panel studies to evaluate further.
- CBC: CBC often shows neutropenia in many X-linked HIGM patients. Apart from neutropenia, anemia, and thrombocytopenia are also sometimes present.
- Quantitative Immunoglobulin Panel: This test helps your doctors to observe the levels of different immunoglobulins (Ig) in the body. In hyper IgM, the levels of IgM are normal or elevated, while other immunoglobulin levels are very low.
Flow Cytometry:
This test involves the study of surface markers, especially CD40L, which is expressed normally on activated T-cells, and is absent in the case of X-linked HIGM type.
If the flow cytometry shows the presence of CD40L, your healthcare specialist will now see the CD40 receptor on the B-cells. A defective or absent CD40 receptor indicates the autosomal recessive form of HIGM.
Functional Assay:
If the flow cytometry reports that CD40L and CD40 receptors are normally expressed and functional, but still there is HIGM, the functional assay will help the doctor to check the B-cell’s class switch recombination (CSR) mechanism.
In a laboratory setting, the pathologist stimulates the B-cells of the suspected individual to see whether these cells switch class from IgM to other immunoglobulins. B-cells of a healthy individual undergo class switching during the procedure. However, if the B-cells fail to switch class even by bypassing the CD40L-CD40 interaction, it suggests AID/ UNG enzyme-deficient HIGM.
Genetic Testing:
Once the above tests suggest the HIGM, genetic testing is the final and confirmatory step done to see the mutations in the genes. If the mutations are in:
- CD40L gene → It is X-linked HIGM
- CD40 receptor gene → It is an autosomal recessive HIGM
- AID/UNG gene → HIGM due to CSR machinery defects
Supportive & Additional Tests:
Even after the confirmation of HIGM, doctors find it useful to do some additional and supportive tests to evaluate the complications caused by the syndrome.
- Liver Function Tests (LFTs) for early liver injuries
- Abdominal Ultrasound & MRI for liver and bile ducts
- Stool Examination for opportunistic infections
- Chest X-ray & CT Scan for recurrent pneumonia and chronic lung damage
- Autoimmune screening for cytopenias
Hyper IgM Syndrome vs. Waldenstrom
A tabular difference between the diseases is given below.
| Feature | Hyper IgM Syndrome (HIGM) | Waldenström Macroglobulinemia (WM) |
|---|---|---|
| Nature of disorder | Primary immunodeficiency (usually genetic | Hematological malignancy (lymphoplasmacytic lymphoma) |
| Cause | Genetic defects affecting CD40L-CD40 interaction or the class-switch recombination mechanism | Acquired somatic mutations (e.g., MYD88 L265P mutation in >90% cases)9Mark D. Ewalt, David Wu, Annette S. Kim, Molecular Hematopathology, Hematopathology, (808-844.e2), (2026).https://doi.org/10.1016/B978-0-443-10940-9.00025-3 |
| Onset | Early in life (infancy or childhood) | Later in life (usually >60 years) |
| Immunoglobulin pattern | Normal/elevated IgM, but very low or absent IgG, IgA, and IgE | Markedly elevated monoclonal IgM, with normal/low IgG and IgA |
| Pathophysiology | Failure of B cells to switch from IgM to other antibody classes due to defective signaling or enzyme function | Uncontrolled clonal proliferation of B lymphocytes producing excess IgM |
| Clinical features | Recurrent bacterial, viral, and opportunistic infections, chronic diarrhea, autoimmune disorders, liver and lung disease | Headache, blurred vision, dizziness, nosebleeds, anemia, neuropathy, enlarged spleen/lymph nodes, increased risk of bleeding |
| Complications | Chronic lung disease, sclerosing cholangitis, increased risk of malignancy (esp. liver cancer, lymphomas) | Hyperviscosity syndrome, bleeding, amyloidosis, secondary immunodeficiency |
| Inheritance/Genetics | Mostly X-linked (CD40L), some autosomal recessive forms | Not inherited. Sporadic somatic mutations |
| Diagnosis | Immunoglobulin profile, flow cytometry, genetic testing, and functional CSR assays | Serum protein electrophoresis and immunofixation, bone marrow biopsy, and MYD88 mutation testing |
| Treatment | Lifelong immunoglobulin replacement therapy, prophylactic antibiotics, and hematopoietic stem cell transplantation | Rituximab-based therapy, chemoimmunotherapy, BTK inhibitors, and plasmapheresis |
Although both diseases appear to be similar by their relatively higher levels of IgM than the other immunoglobulins, both are totally different with cause, symptoms, and treatment.
Treatment of Hyper IgM Syndrome
Once the diagnosis is established, the next step is the management and treatment of the disease, which involves the replacement of missing antibodies, preventing infections, and managing complications of HIGM.
Immunoglobulin Replacement Therapy:
To compensate for the immunoglobulin in the HIGM patient, doctors give the antibodies directly into the patient’s body. Although this treatment option lowers the frequency and severity of bacterial infection, it is not very effective against opportunistic diseases.
The immunoglobulin replacement therapy can be done by either:
- Intravenous immunoglobulin (IVIG): Given every 3 to 4 weeks in many patients.
- Subcutaneous immunoglobulin (SCIG): Administered more frequently (e.g., weekly), often used when venous access is problematic or to maintain more stable IgG levels.
Infection Prophylaxis & Management:
As the patient becomes highly vulnerable to different infections by HIGM, the prophylaxis and management for different infections can be a life-saving step. Trimethoprim-sulfamethoxazole (TMP-SMX) is considered one of the most important measures for the prophylaxis of PCP infection, a very common infection in HIGM patients.
Primary prophylaxis in HIGM focuses on PCP prevention with trimethoprim-sulfamethoxazole. Routine systemic antifungal or antiviral prophylaxis is not universally recommended and is used only when individual risk factors (transplant, persistent neutropenia, active CMV) are present.
Neutropenia Management:
Your doctor may suggest the Granulocyte Colony-Stimulating Factor (G-CSF) method to manage the neutropenia, in which the production of neutrophils is stimulated.
Hematopoietic Stem Cell Transplantation (HSCT):
HSCT is the only curative procedure for the hyper-immunoglobulin M syndrome, especially for the X-linked and AR types. It involves replacing the patient’s defective immune system with the hematopoietic stem cells of the healthy donor. HSCT retains the normal B and T-cell interaction, allowing the immunoglobulin class switching.
Although the HSCT has the best outcomes when performed early, its outcomes vary by center, donor match, and liver condition.
Gene Therapy:
Gene therapy is a promising future treatment for HIGM syndrome, in which the patient’s own hematopoietic stem cells are genetically corrected to reestablish a functional immune system. Currently, this procedure is in its experimental phase.
Life Expectancy of Hyper IgM Syndrome
The life expectancy of HIGM depends on how early it is diagnosed.
- Without treatment, the recurrent and opportunistic infections may lead to complications like chronic lung disease, liver cirrhosis, and malignancies, which definitely shorten the life span of the patient.
- With modern care, like immunoglobulin replacement therapy and prophylaxis, the patients live well into adulthood, but the quality of life may still be affected due to infections.
- With HSCT performed early in the life of the patient, the disease can be cured and bring the life expectancy near normal.
Conclusion
Hyper IgM syndrome is a heterogeneous group of primary immunodeficiencies where defective class switch recombination leads to excess IgM and deficiency of other antibody classes. It is caused by either the failure of CD40L-CD40 interaction or the deficiency of enzymes like AID or UNG for the class switch recombination, both are steps important for switching the class from IgM to other immunoglobulin productions.
HIGM presents with recurrent diseases with opportunistic infections, leading to different complications like chronic diarrhea, sclerosing cholangitis, liver failure, and neutropenia. It is treated with immunoglobulin replacement therapy, infection prophylaxis, and hematopoietic stem cell replacement methods after the diagnosis. Through flow cytometry, functional assay, and gene therapy are useful methods in the diagnosis of the disease.
References
[1] Dunn CP, de la Morena MT. X-Linked Hyper IgM Syndrome. 2007 May 31 [Updated 2020 Feb 20]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1402/.
[2] Patel P, Jamal Z, Ramphul K. Immunoglobulin. [Updated 2023 Aug 28]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK513460/
[3] Aziz M, Iheanacho F, Hashmi MF. Physiology, Antibody. 2023 May 1. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 31536276.
[4] Liu JC, Zhang K, Zhang X, Guan F, Zeng H, Kubo M, Lee P, Candotti F, James LK, Camara NOS, Benlagha K, Lei JH, Forsman H, Yang L, Xiao W, Liu Z, Liu CH. Immunoglobulin class-switch recombination: Mechanism, regulation, and related diseases. MedComm (2020). 2024 Aug 13;5(8):e662. doi: 10.1002/mco2.662. PMID: 39144468; PMCID: PMC11322596.
[5] Cerutti, A., Zan, H., Kim, E. C., Shah, S., Schattner, E. J., Schaffer, A., & Casali, P. (2002). Ongoing In Vivo Immunoglobulin Class Switch DNA Recombination in Chronic Lymphocytic Leukemia B Cells.Journal of Immunology (Baltimore, Md.: 1950),169(11), 6594. https://doi.org/10.4049/jimmunol.169.11.6594
[6] Tieu, J., Smith, R. M., Gopaluni, S., Kumararatne, D. S., McClure, M., Manson, A., Houghton, S., & W Jayne, D. R. (2021). Rituximab Rituximab-associated hypogammaglobulinemia in Autoimmune Disease.Frontiers in Immunology,12, 671503. https://doi.org/10.3389/fimmu.2021.671503
[7] Rodríguez C, Carrión F, Marinovic MA, Chávez E, Preisler J, Pooley F, Futatani T, Ochs HD. Síndrome de hiper-IgM asociado a colangitis esclerosante y neoplasia vesicular: caso clínico [X-linked hyper-IGM syndrome associated with sclerosing cholangitis and gallbladder neoplasm: clinical case]. Rev Med Chil. 2003 Mar;131(3):303-8. Spanish. PMID: 12790080.
[8] O’Gorman MR. Measurement of CD40 ligand (CD154) expression on resting and in vitro-activated T cells. Curr Protoc Cytom. 2001 May;Chapter 6:Unit 6.7. doi: 10.1002/0471142956.cy0607s12. PMID: 18770718.
[9] Mark D. Ewalt, David Wu, Annette S. Kim, Molecular Hematopathology, Hematopathology, (808-844.e2), (2026).https://doi.org/10.1016/B978-0-443-10940-9.00025-3
[10] Davies, E. G., & Thrasher, A. J. (2010). Update on the hyperimmunoglobulin M syndromes.British Journal of Haematology,149(2), 167. https://doi.org/10.1111/j.1365-2141.2010.08077.x
[11] Dunn CP, de la Morena MT. X-Linked Hyper IgM Syndrome. 2007 May 31 [Updated 2020 Feb 20]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Table 4. [Treatment of Manifestations in Individuals with X-Linked Hyper IgM Syndrome]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1402/table/xlhi.T.treatment_of_manifestations_in_in/
[12] CETEAN, S., CĂINAP, C., CONSTANTIN, M., CĂINAP, S., GHERMAN, A., OPREAN, L., HANGAN, A., & OPREAN, R. (2015). The importance of the granulocyte-colony stimulating factor in oncology. Clujul Medical, 88(4), 468. https://doi.org/10.15386/cjmed-531
[13] Al-Saud B, Al-Jomaie M, Al-Ghonaium A, Al-Ahmari A, Al-Mousa H, Al-Muhsen S, Al-Seraihy A, Arnaout R, Elshorbagi S, Al-Dhekri H, Ayas M. Haematopoietic stem cell transplant for hyper-IgM syndrome due to CD40 defects: a single-centre experience. Bone Marrow Transplant. 2019 Jan;54(1):63-67. doi: 10.1038/s41409-018-0219-0. Epub 2018 Jun 8. PMID: 29884852.
[14] Dunn CP, de la Morena MT. X-Linked Hyper IgM Syndrome. 2007 May 31 [Updated 2020 Feb 20]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1402/