
{kind=link}

G6PD Deficiency Overview G6PD deficiency is the most common human enzyme deficiency worldwide and is inherited in an X-linked pattern, affecting males more frequently. The condition impairs the ability of red blood cells to defend themselves against oxidative stress, resulting in low haemoglobin oranemia. All bodily cells have the G6PD enzyme. It acts as a housekeeper to shield cells from damage done by reactive oxygen species (ROS). Being oxygen carriers, erythrocytes are particularly vulnerable because, unlike other cells, they cannot regenerate new enzyme once mature.[1] In G6PD deficiency, oxidative stress, triggered by infections, certain drugs (such as antimalarials, sulfonamides, and some antibiotics), or fava bean ingestion (favism), can lead to premature destruction of red blood cells, resulting in hemolytic anemia. Symptoms may include fatigue, jaundice, dark urine, and in newborns, severe jaundice.
How common is it?: G6PD Deficiency Overview
The disease is quite common, affecting approximately 400 million people worldwide.[2] Compared to the rest of the world, the condition is more prevalent in some areas of Africa, the Middle East and the Mediterranean. Interestingly, a majority of people remain asymptomatic. They often are unaware of the disease until they experience an episode of hemolysis following a trigger. However, later on, they can still live a normal life once they know what to avoid.
Causes of G6PD Deficiency
G6PD deficiency is an X-linked recessive disorder. It means the locus, i.e., home of the gene that codes for G6PD, is on the X chromosome. As males, are XY, they only get one copy of the X chromosome, which is from their mothers. In contrast, females are XX and get one copy from both their mother and father. That’s why, for the disease to manifest in females, it is possible only if they inherit both defective copies, which is very rare. Hence, males are usually the ones with the disease. However, due to X-chromosome inactivation (Lyonization), some heterozygous females may also show symptoms ranging from mild to significant hemolysis. Females with one defective gene often act as carriers and can pass the condition to their children.
Types of G6PD Deficiency
Different variants of the G6PD gene result in varying levels of enzyme activity. Interestingly, in some cases, enzyme activity is seen to be even increased above normal. The World Health Organisation (WHO) classifies the disease into 5 classes based on the level of enzyme activity and the spectrum of symptoms. These are:
| Class | Enzyme Activity | Clinical Symptoms | Example Variants |
|---|---|---|---|
| Class I | <10% | Severe deficiencywith chronic non-spherocytic hemolytic anemia (CNSHA) | Rare (e.g., G6PD Harilaou) |
| Class II | <10% | Severe deficiency, but hemolysis occurs only with stress (e.g., infections, drugs, fava beans) | G6PD Mediterranean, G6PD Canton |
| Class III | 10-60% | Moderate deficiency; episodic hemolysis | G6PD A− (common in Africa) |
| Class IV | >60% | Normal activity; no clinical symptoms | G6PD B (normal variant) |
| Class V | >150% | Increased enzyme activity; not clinically significant | Rare |
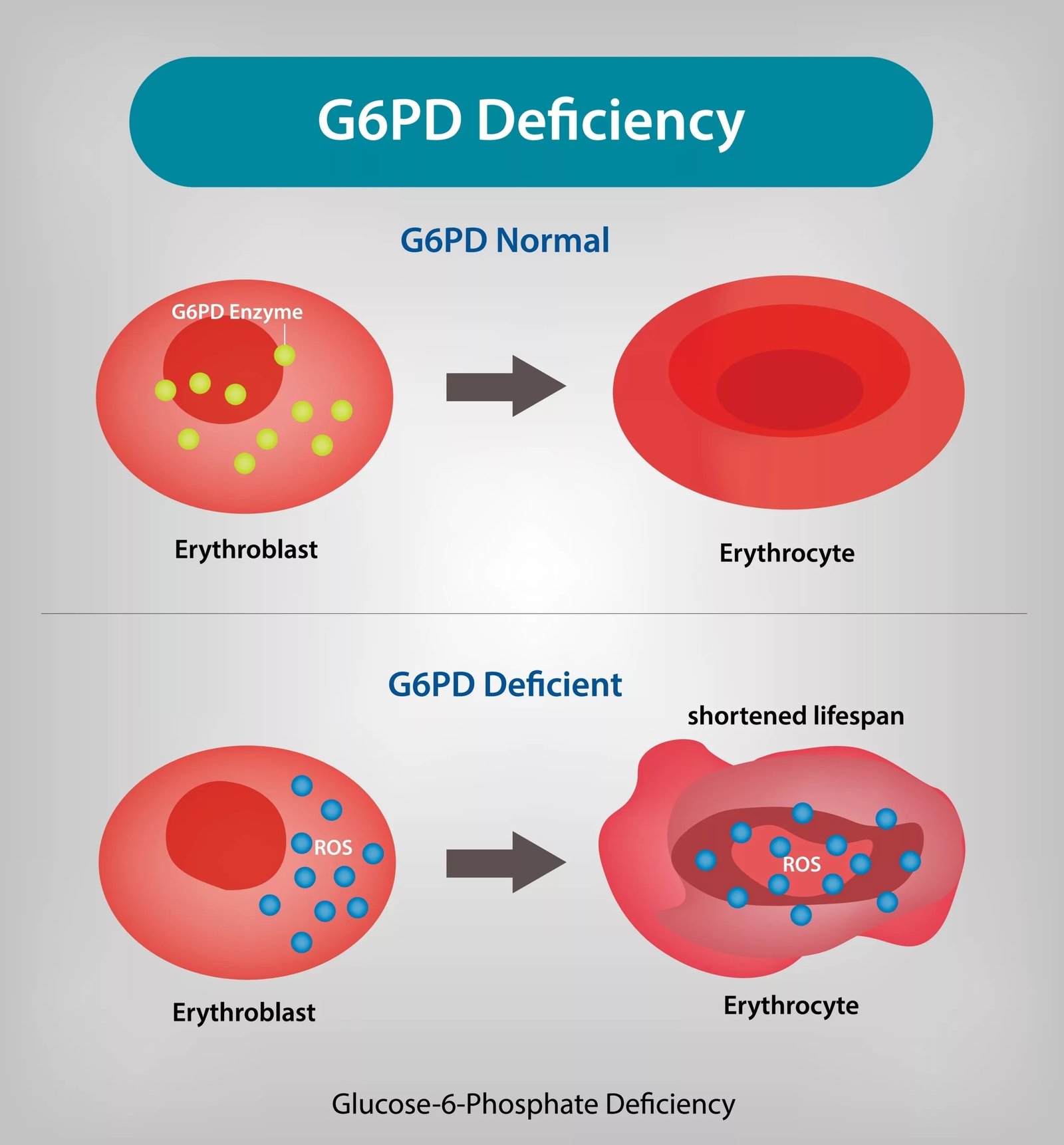
G6PD Deficiency Triggers
The most well-known trigger is ingestion of fava beans. Other triggers include:
Medications:
Some drugs particularly trigger the hemolysis in G6PD-deficient people. These include.
- Antimalarials: Primaquine, Tafenoquine (chloroquine is usually safe in most variants)
- Sulfa Drugs: Sulfonamides, Sulfasalazine
- Antibacterials: Nitrofurantoin, Chloramphenicol, Dapsone
- Others: Even aspirin in large doses can trigger the breakdown of red blood cells.
Infections:
When the body is fighting any viral or bacterial infection, such as the flu or pneumonia, red blood cells (RBCs) end to be at a higher risk of breaking down. In addition, some conditions like diabetic ketoacidosis (DKA) are also notable triggers.hemolytic anemia.Eur J Pediatr. 2008 Dec. 167(12):1435-9.” style=”position:relative;color:#309b65;cursor:help;border-bottom:1px dotted #309b65;font-weight:bold”>[3]
Environmental Factors:
Chemicals such as naphthalene (found in mothballs) and henna have been reported to trigger hemolysis in susceptible individuals.
G6PD Deficiency Symptoms
Most patients with G6PD deficiency are asymptomatic. After 24 hours of birth, neonatal jaundice may be seen[4], but it is not a significant finding, and jaundice subsides within a few weeks. However, in some infants, excessive injury to red blood cells occurs, and the product of breakdown, called bilirubin, can lead to brain damage (kernicterus). These cases need exchange transfusion.
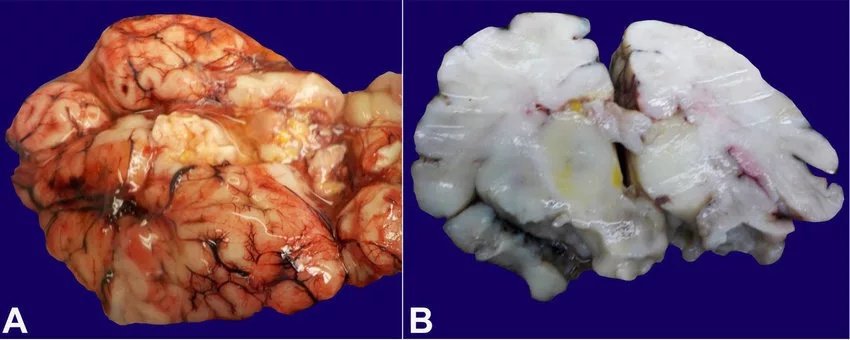
(Kernicterus. A -Gross image of the brain with bilirubin pigment deposition in the fresh state; B -Gross image of coronal section of the brain with bilirubin pigment deposition in the basal ganglia, post-fixation. Image courtesy:Researchgate, available via CC 4.0 International)
Adults are not usually anemic. But after exposure to the trigger, hemolysis crisis may begin. This process takes 48-72 hours after the exposure to oxidation stress. When having hemolysis, the patient may present with symptoms such as:
- Weakness
- Tachycardia or increased heart rate above normal
- Jaundice or yellowing of skin
- Hematuria or blood in urine, causing dark colour of urine
- Upper quadrant pain
Physical examination, an enlarged spleen, i.e spleenomegaly, may be present.[5] Rarely, skin sores may also be visible.
These symptoms develop fast, and hence we call it a hemolytic crisis. This stage lasts 8-14 days and typically resolves on its own.
G6PD Deficiency Diagnosis
If a patient shows signs of anemia, the doctor will usually start by taking a complete medical history and then doing a physical examination. The doctor will also ask about any kind of allergy, infection or recent change of medication. Later, they will order some tests such as:
Complete Blood Count (CBC):
To check the haemoglobin level, mean corpuscular volume (average size of red blood cell) and red cell distribution width.
Peripheral Smear:
To check for shapes and detect abnormal cells and reticulocyte count. A frequent finding is the presence of Heinz bodies (denatured haemoglobin) or bite cells in the peripheral smear of patients with G6PD deficiency. Reticulocyte count is increased in cases of hemolytic anemia. The increased reticulocyte count is the bone marrow response to compensate for increased red cell breakdown.
Serum Bilirubin:
To check the level of direct and indirect bilirubin. It is a waste product of red blood cells.
In some cases, doctors may recommend some other tests, such as Urinalysis for hematuria, haptoglobinlevels and detection of urinary hemosiderin. These markers indicate severe hemolysis.

Specific tests to diagnose G6PD Deficiency:
In cases where a pattern of hemolysis raises suspicion of G6PD deficiency and there is a suggestive family history or in prevalent areas, doctors recommend screening tests for G6PD deficiency.[6] If a screening test comes positive, it should be confirmed by a quantitative test. Standard tests include:
Beutler test: A rapid fluorescent spot test that detects nicotinamide adenine dinucleotide phosphate (NADPH). NADPH fluoresces when it forms. The absence of fluorescence suggests the enzyme is deficient. This test is widely used but less reliable in females due to X-linked inheritance..[7]
Quantitative assay of G6PD activity: It is the standard test to check levels of G6PD activity by using a spectrophotometer. Limitations include reduced accuracy during or soon after an acute hemolytic episode, as young red cells may show normal activity.
Cytochemical or chrome inhibition test: It includes looking at individual RBCs under a microscope using special dyes or lasers to see how many cells are G6PD deficient. It is not very common and not advised in routine. However, it is more sensitive in detecting mosaic pattern (presence of abnormal cells between normal cells) in females.
Genetic testing: These are useful in cases when enzyme assays are inconclusive or to detect carrier status or a specific G6PD gene variant.
G6PD Deficiency Treatment
Most people with G6PD deficiency don’t need treatment. However, their healthcare provider will advise them to avoid certain drugs, chemicals and fava beans, as they can lead to oxidative stress. The crucial part of management is the identification and removal of the precipitating substance.
Management of Acute Hemolytic Episode:
As hemolysis is self-limited and resolves itself in 8-14 days, only supportive management is sufficient in most cases. The management includes ensuring hydration and monitoring of different parameters such as bilirubin, LDH, renal function tests and complete blood count over the course of a few days. However, in rare cases, such as in patients with severe anemia (Hb <7g/dl) or renal failure, transfusions may be indicated.
Neonatal Jaundice Management:
If jaundice is evident in the newborn, the first-line treatment is phototherapy. In some cases, despite phototherapy, the bilirubin level continues rising, consequently putting the baby at the risk of kernicterus(bilirubin-induced brain damage). These babies require transfusions.[8]
Chronic non-spherocytic Hemolytic Anemia:
Some G6PD variants can cause ongoing hemolysis. Their management includes daily folic acid supplementation and monitoring of Complete blood count and bilirubin levels. In addition, ultrasonography of the abdomen is also indicated from time to time to check for the formation of gallstones, made up of bile.
Role of Splenectomy:
A typical approach to hemolytic anemia, splenectomy is not found to be beneficial in this disease, as red blood cell destruction occurs because of oxidative stress.
Differential diagnosis of G6PD Deficiency
Some diseases which cause low haemoglobin levels may mimic the symptoms of G6PD deficiency. These include:
- Autoimmune hemolytic anemia
- Hereditary spherocytosis
- Sickle cell anemia
- Thalassemia
- Methemoglobinemia
Conclusion
G6PD deficiency is a commonly occurring enzyme deficiency which can lead to low levels of haemoglobin. Although it is not lethal and patients can have a normal life despite having been diagnosed with it, sometimes severe hemolytic anemia may occur because of the extensive breakdown of red blood cells. In endemic areas or suspected people, it is essential to rule out the diseases so further hemolytic attacks may be prevented. The disease is treatable and usually resolves itself with supportive management; however, some cases may need transfusions.
References
[1] [Guideline] Guide to G6PD deficiency rapid diagnostic testing to support P. vivax radical cure. World Health Organization.
[2] Nkhoma ET, Poole C, Vannappagari V, Hall SA, Beutler E. The global prevalence of glucose-6-phosphate dehydrogenase deficiency: a systematic review and meta-analysis.Blood Cells Mol Dis. 2009 May-Jun. 42(3):267-78.
[3] Ozbay Hosnut F, Ozcay F, Selda Bayrakci U, Avci Z, Ozbek N. Etiology of hemolysis in two patients with hepatitis A infection: glucose-6-phosphate dehydrogenase deficiency or autoimmune hemolytic anemia.Eur J Pediatr. 2008 Dec. 167(12):1435-9.
[4] Isa HM, Mohamed MS, Mohamed AM, Abdulla A, Abdulla F. Neonatal indirect hyperbilirubinemia and glucose-6-phosphate dehydrogenase deficiency.Korean J Pediatr. 2017 Apr. 60 (4):106-111.
[5] Prchal JT, Gregg XT. Red cell enzymes.Hematology Am Soc Hematol Educ Program. 2005. 19-23.
[6] Minucci A, Giardina B, Zuppi C, Capoluongo E. Glucose-6-phosphate dehydrogenase laboratory assay: How, when, and why?.IUBMB Life. 2009 Jan. 61(1):27-34.
[7] Beutler E. G6PD deficiency.Blood. 1994 Dec 1. 84(11):3613-36.
[8] Murki S, Dutta S, Narang A, Sarkar U, Garewal G. A randomized, triple-blind, placebo-controlled trial of prophylactic oral phenobarbital to reduce the need for phototherapy in G6PD-deficient neonates.J Perinatol. 2005 May. 25(5):325-30.