
 is a rare hereditary cancer syndrome that dramatically increases a person’s lifetime risk of...){kind=link}

Li Fraumeni Syndrome Li-Fraumeni syndrome (LFS) is a rare hereditary cancer syndrome that dramatically increases a person’s lifetime risk of developing multiple types of cancer. Dr. Frederick Li and Dr. Joseph Fraumeni identified this syndrome in 1969 when they noticed unusual patterns of cancer in some families. Their research found that they had an inherited tendency for cancer, which is now known as one of the most essential familial cancer syndromes.[1]
This rare genetic disorder affects about 1 in 5,000 to 1 in 20,000 people globally. But its effect on the affected families is very hurtful because the inherited cancer predisposition passes to several generations, without leaving anyone. The defining feature of the syndrome is the multiple primary tumors occurring in some individuals. Other family members develop different cancers at much younger ages.
With modern genetic testing, an increasing number of people are diagnosed with LFS earlier in life and can make a knowledgeable decision regarding health, lifestyle, and family planning. Although frightening, the condition still offers hope as researchers develop new screening techniques and preventive measures.[2]
Genetic Basis & Causes of Li-Fraumeni Syndrome: Li Fraumeni Syndrome
Li-Fraumeni syndrome involves a major genetic defect at the most fundamental level in the TP53 gene, located on chromosome 17. The TP53 gene is one of the most critical tumor suppressor genes, controlling cell growth, DNA repair, and programmed cell death (apoptosis). In a healthy condition, the TP53 gene acts as a “guardian of the genome,” preventing damaged cells of the body from becoming cancerous.[3]
When TP53 mutates, it loses its ability to protect against abnormal cell growth. The modified gene is no longer able to regulate cell division efficiently or even inhibit the abnormal growth of cells, thereby providing the environment that allows the development of tumors. When TP53 is mutated, its ability to prevent abnormal cell growth is lost, allowing malignant transformation. In LFS, an affected person inherits one mutated copy of the gene from a parent. Cancer typically develops after a “second hit” occurs in the remaining normal copy within certain cells. This makes LFS an autosomal dominant condition with high penetrance. While most individuals with LFS inherit the mutation, some cases arise from new (de novo) TP53 mutations.
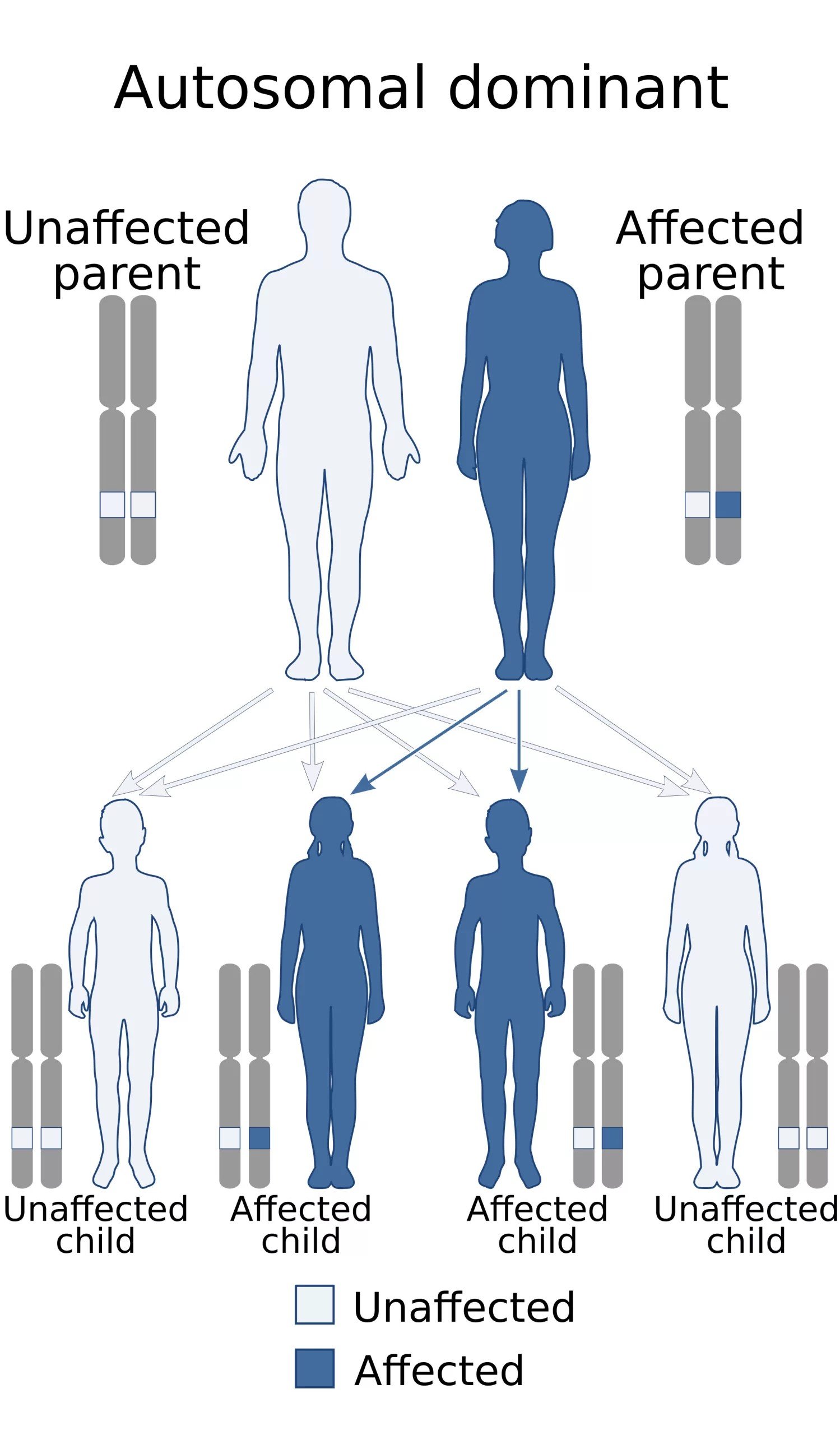
Autosomal dominant inheritance pattern. Credit: Wikimedia Commons, CC BY-SA 4.0, viaWikipedia.
Cancer Types and Symptoms
Li-Fraumeni syndrome is a condition that leads you to live in fear of getting cancer. LFS is one of the most complex cancer syndromes because it predisposes to a vast range of malignancies. The disease has been linked explicitly to certain types of cancer that comprise the LFS tumor spectrum, but any kind of cancer is possible in an affected person.
What cancers are associated with Li-Fraumeni Syndrome?
- Soft-tissue sarcomas are one of the most typical LFS-related malignancies that appear in the muscles, fat, blood vessels, nerves, and joints. Although these are rare cancers, they tend to develop in childhood or young adulthood and may occur in abnormal locations within the body.
- Osteosarcomas develop in bone cells and can also occur during the teenage years, when bone growth is most active.
- Studies indicate that the lifetime risk of cancer among individuals with LFS is 90% among women and approximately half in men, primarily because premenopausal breast canceris common in women. The age of onset is significantly younger than that of the general population, and several LFS-related cancers present before age 30 or 45.
- Brain tumours, both childhood brain tumours and adult gliomas, as well as the rare choroid plexus tumor.
- Adrenocortical tumors arise in the adrenal gland located above each kidney, and they are found in the LFS family with unusual frequency. Such rare cancers secrete excessive amounts of adrenal hormones such as cortisol, testosterone, and other steroid substances and cause other health problems, beyond the cancer itself.
- In Li-Fraumeni syndrome, there is also an increased incidence of acute leukemias, especially acute lymphoblastic leukemia in children.
Radiation Risks and Secondary Malignancies
- Radiation exposure, often used in standard cancer treatment, can increase the risk of radiation-induced cancers in LFS patients. This treatment creates challenges for physicians, who must carefully monitor the risks and benefits of traditional cancer therapies. Instead, surveillance protocols focusing on early detection and preventive screening play a larger role in management.
- Other secondary malignancies that occur with LFS are:
The risk of contracting many primary tumors in the course of life is one of the most challenging issues of the syndrome, as people who have survived one cancer are at risk of getting another malignancy very soon.[4]
What are the symptoms of Li-Fraumeni Syndrome?
It is essential to know that LFS does not itself have any symptoms. Instead, it causes the symptoms of different cancers. That is why surveillance is necessary. But people and families are advised to be vigilant about:[5]
- Unexplained lumps or swelling (potential sarcoma).
- Constant headaches, nausea, vomiting, or eye problems (possible brain tumor).
- Night sweats, unexplained weight loss, and fatigue.
- Pain in the bones or bone fractures (possible osteosarcoma).
- Abnormal bleeding or bruising (possibly leukemia).
- Alteration in a mole or epidermal lesion (possible melanoma).
- Any other chronic or abnormal medical complaint.
Due to the risk of cancer, Doctors should assess any new or persistent symptoms and be familiar with LFS at the earliest.
Cancer Risk By Age & Gender:
Li-Fraumeni syndrome age patterns indicate the especially devastating effects of the disease on young adults and children. Although cancer may occur at any age in an afflicted person, its peak incidence is much earlier than in the general population. LFS women may be at increased risk of cancer throughout life relative to men, in part due to the significant component of breast cancer risk.[6]
Diagnostic Criteria & Clinical Classifications
Li-Fraumeni syndrome can only be diagnosed with careful evaluation of family history patterns, types of cancer, and age of onset of different cancers in several generations. Medical professionals use diagnostic criteria to identify families with TP53 mutations, which can benefit from genetic testing.
Classic Li-Fraumeni Criteria:
According to the classic Li-Fraumeni criteria, invented by the discoverers of the syndrome in 1969, the proband (first affected family member) has sarcoma before 45 years of age, plus one first-degree family member diagnosed with any cancer before 45 years of age, plus one first- or second-degree relative with any tumor before 45 years of age or sarcoma at any age. Although specific, these initial criteria failed to identify some of the families with TP53 mutations.[7]
Chompret Criteria:
A more recent development, the Chompret criteria, broadened the diagnostic framework to include more families at risk. These include individuals aged 46 or younger with LFS spectrum tumors, with multiple primary tumors of the LFS spectrum, as well as individuals with adrenocortical carcinoma or choroid plexus tumors, either with or without family history. Chompret criteria define approximately 20% of families carrying TP53 mutations who would otherwise not fit classic criteria.[8]
Li-Fraumeni-like Syndrome (LFL):
Li-Fraumeni-like syndrome (LFL) is used to categorize families with some LFS-like characteristics but not all of the requirements. The Birch definition needs at least one proband who had cancer in childhood or particular types of adult cancers before age 45, along with typical LFS cancers in family members, and cancer in a relative before age 60. The Eeles definition only involves two relatives with malignancies related to LFS at any age.[9]
How is Li-Fraumeni Syndrome Diagnosed?
To confirm Li-Fraumeni syndrome, genetic tests use direct DNA sequencing of the TP53 gene. The first step in the testing process is to obtain a sample of blood or saliva from an affected person. Laboratory technicians isolate the DNA, and both copies of the TP53 gene are analyzed against the standard reference sequences.[10]
Blood sample for TP53 (tumor protein 53) mutation analysis testing, to determine risk of certain types of cancers. Li-Fraumeni syndrome.
Genetic Testing:
DNA sequencing technology has the potential to detect all forms of TP53 mutations, including point mutations, deletions, insertions, and more significant genomic rearrangements. As labs identify a mutation, genetic counselors examine existing databases to determine whether the particular change has been previously linked to Li-Fraumeni syndrome. Certain genetic variations are clearly pathogenic mutations, while others are variants of uncertain importance that need further research.[11]
Cascade testing can be performed in a family once positive results of a TP53 mutation have been confirmed by positive genetic tests. After the laboratories have detected the specific mutation in a family, the rest of the family members can undergo particular testing to establish their mutation status. This is less expensive and more reliable than sequencing the entire genome of each person.
The decision to undergo genetic testing should be accompanied by clear, detailed genetic counseling to address the benefits, limitations, and potential outcomes of the results. Cancer surveillance, family planning, medical management, psychological stress, insurance issues, and family relationship issues can all be valuable information obtained through testing, but can also generate psychological stress.
Is there any Treatment for Li-Fraumeni Syndrome?
Treatment for LFS is not a treatment for the syndrome itself, but for the individual cancers a patient develops. Management is highly personalized and must consider the high risk of secondary cancers.
- Surgical Removal: The primary treatment for many solid tumors (e.g., sarcomas, breast cancer, brain tumors) is surgical resection with wide margins.[12]
- Radiation Therapy: Used with extreme caution. Due to the defective DNA repair mechanism, radiation significantly increases the risk of secondary malignancies in LFS patients. It should be used in the most serious, life-saving cases where we have no other options.
- Chemotherapy: Standard chemotherapy drugs are used, although oncologists might prefer chemotherapy regimens with less long-term toxicity.
- New Therapies: Research on the mutant p53 protein and the specific vulnerabilities of p53-deficient cells is ongoing. These include:
- The Role of a Tumor Board: Management of LFS-related cancers is best done by a multi-disciplinary team (MDT) including oncologists, geneticists, surgeons, and radiologists who can weigh the risks and benefits of each treatment option.[13]
Cancer Screening and Surveillance Protocols
For families with LFS, early detection is the cornerstone of survival. Since cancer risk is so high, doctors recommend lifelong surveillance. These protocols aim to catch tumors in their earliest stages.
A standard surveillance protocol may include:
- Total body MRI – to scan for soft-tissue sarcomas, bone tumors, and hidden cancers.
- Brain MRI – for early detection of brain tumors and choroid plexus tumors.
- Mammogram and breast MRI – especially for women at risk of premenopausal breast cancer.
- Abdominal ultrasound – to screen for tumors in the adrenal gland, kidneys, and liver.
- Colonoscopy – to detect early colon cancer and polyps.
- Urinalysis and complete blood count – to check for leukemias and kidney issues.
- Neurologic and skin examinations – for melanoma, nerve-related tumors, and brain cancers.[14]
In addition to imaging, blood chemistries and hormone tests (such as cortisol, testosterone, and adrenal hormones) may be included to identify endocrine-related cancers early.
Unlike the general population, where screenings typically begin after age 40 or 50, LFS surveillance often starts in childhood. For example, children may receive annual whole-body MRIs or ultrasounds.[15]
The goal is to find cancers before symptoms appear, giving patients the best chance of successful treatment. While this constant monitoring can feel burdensome, many families find comfort in knowing that surveillance increases the odds of survival and peace of mind.
Lifestyle Modifications and Preventive Measures
Although you cannot remove genetic predisposition, lifestyle changes can decrease the risk of cancer and enhance overall health outcomes in patients with Li-Fraumeni syndrome.[16]
Some of the most important preventive steps include:
- A healthy lifestyle, characterized by the consumption of nutrient-rich foods rich in antioxidants, may support DNA repair and enhance the immune system. Regular physical activity, when medically suitable, can help achieve better health and possibly reduce the risk of cancer.
- Sun protection: Since melanoma risk is elevated, limiting sun exposure and using sunscreen is crucial. Wear high SPF broad-spectrum sunscreen, protective clothing, and wide-brimmed hats to protect yourself from the sun’s harmful rays. Avoid peak sun hours and seek shade whenever possible. Determine these habits early in the lives of affected children.
- Avoiding tobacco and alcohol: Never smoke, avoid secondhand smoke; tobacco is a significant risk factor in the presence of cancers already elevated in LFS. Minimize alcohol intake, which contributes to some types of cancer.
- Minimizing radiation exposure: This is crucial due to the increased risk of radiation-related cancers. Yet, it must also be weighed against the value of medical imaging as a surveillance mechanism. Requires a thorough discussion with clinicians regarding the most effective screening plans.
Future Family Planning: Options for Parents with a TP53 Mutation
For individuals with a TP53 mutation who are considering having children, genetic counseling is essential to understand the inheritance pattern and discuss the available options to reduce the risk of passing the syndrome to their offspring. A couple with a TP53 mutation has a 50% probability of passing the mutation to their children. Some reproduction choices can guide them:[17]
- Prenatal Testing: To prepare the family to deal with the situation medically or to consider terminating the pregnancy, doctors can test the developing baby to determine whether it has inherited certain conditions, using techniques such as chorionic villus sampling (in the first trimester) or amniocentesis (in the second trimester).[18]
- Preimplantation Genetic Diagnosis (PGD) in IVF enables the selection of healthy embryos before pregnancy. It involves medical intervention and is very expensive, but it promises genetically unaffected children.
- Other Alternatives: The couples can opt for natural conception, in which they have proper surveillance for the ill children, adoption, donor gametes, or be left childless.
Conclusion
Li-Fraumeni syndrome is one of the most essential hereditary cancer syndromes in medicine that poses some huge problems for the affected families. The syndrome linked to mutations in TP53 has contributed to the understanding of this tumor suppressor gene in cancer development. Though the risks of cancer are still overwhelming, a well-developed surveillance plan, behavioral change, and medical developments can provide viable management options.
Genetic counseling supports informed decisions about testing, family planning, and medical care. It is possible only through the early detection of treatable cancers through appropriate screening. Progress in research on new treatment options, better disease surveillance, and potential prevention strategies promises a brighter future.
It takes cooperation between individuals with the syndrome, their families, health practitioners, genetic counseling professionals, and researchers to achieve success, and the medical community never stops striving to make life better and easier for people living with this relatively uncommon but noteworthy hereditary cancer syndrome.
References
[1] Li, F. P., & Fraumeni, J. F. (1969). Soft-tissue sarcomas, breast cancer, and other neoplasms: A familial syndrome?Annals of Internal Medicine, 71(4), 747–752.
[2] Malkin, D. (2011). Li-Fraumeni syndrome.Genes & Cancer, 2(4), 475–484.
[3] Bougeard, G., Renaux-Petel, M., Flaman, J. M., Charbonnier, C., Fermey, P., Belotti, M., … & Frebourg, T. (2015). Revisiting Li-Fraumeni syndrome from TP53 mutation carriers.Journal of Clinical Oncology, 33(21), 2345–2352.
[4] Malkin, D., Li, F. P., Strong, L. C., Fraumeni, J. F., Nelson, C. E., Kim, D. H., … & Friend, S. H. (1990). Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms.Science, 250(4985), 1233–1238.
[5] Bougeard, G., Renaux-Petel, M., Charbonnier, C., Fermey, P., Belotti, M., Gauthier-Villars, M., … & Frebourg, T. (2015). Revisiting Li-Fraumeni syndrome: clinical and molecular spectrum in 91 families.Journal of Clinical Oncology, 33(21), 2345–2352.
[6] Mai, P. L., Best, A. F., Peters, J. A., DeCastro, R. M., Khincha, P. P., Loud, J. T., … & Savage, S. A. (2016). Risks of first and subsequent cancers among TP53 mutation carriers in the National Cancer Institute Li-Fraumeni syndrome cohort.Cancer, 122(23), 3673–3681.
[7] .Li, F. P., & Fraumeni, J. F. (1969). Soft-tissue sarcomas, breast cancer, and other neoplasms: A familial syndrome?Annals of Internal Medicine,
[8] Chompret, A., Abel, A., Stoppa-Lyonnet, D., Brugières, L., Pagès, S., Feunteun, J., & Bonaiti-Pellie, C. (2001). Sensitivity and predictive value of criteria for p53 germline mutation screening.Journal of Medical Genetics, 38(1), 43–47.
[9] Birch, J. M., Hartley, A. L., Tricker, K. J., Prosser, J., Condie, A., Kelsey, A. M., … & Evans, D. G. (1994). Prevalence and diversity of constitutional mutations in the p53 gene among 21 Li-Fraumeni families.Cancer Research, 54(5), 1298–1304.
[10] Gonzalez, K. D., Noltner, K. A., Buzin, C. H., Gu, D., Wen-Fong, C. Y., Nguyen, V. Q., … & Weitzel, J. N. (2009). Beyond Li-Fraumeni syndrome: clinical characteristics of families with p53 germline mutations.Journal of Clinical Oncology, 27(8), 1250–1256.
[11] Tinat, J., Bougeard, G., Baert-Desurmont, S., Vasseur, S., Martin, C., Bouvignies, E., … & Frebourg, T. (2009). 2009 version of the Chompret criteria for Li-Fraumeni syndrome.Journal of Clinical Oncology, 27(26), e108–e109.
[12] Villani, A., Shore, A., Wasserman, J. D., Stephens, D., Kim, R. H., Druker, H., … & Malkin, D. (2016). Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: A prospective observational study.The Lancet Oncology, 17(9), 1295–1305.
[13] Masciari, S., Dewanwala, A., Stoffel, E. M., Lauwers, G. Y., Zheng, H., Achatz, M. I., … & Garber, J. E. (2011). Gastric cancer in individuals with Li-Fraumeni syndrome.Genetics in Medicine, 13(7), 651–657.
[14] .Villani, A., Tabori, U., Schiffman, J., Shlien, A., Beyene, J., Druker, H., … & Malkin, D. (2011). Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: a prospective observational study.The Lancet Oncology, 12(6), 559–567.
[15] .Kratz, C. P., Achatz, M. I., Brugières, L., Frebourg, T., Garber, J. E., Greer, M. C., … & Savage, S. A. (2017). Cancer screening recommendations for individuals with Li-Fraumeni syndrome.Clinical Cancer Research, 23(11), e38–e45.
[16] Ballinger, M. L., Best, A., Mai, P. L., Khincha, P. P., Loud, J. T., Peters, J. A., … & Savage, S. A. (2017). Baseline surveillance in Li-Fraumeni syndrome using whole-body magnetic resonance imaging: a meta-analysis.JAMA Oncology, 3(12), 1634–1639.
[17] Gonzalez, K. D., Noltner, K. A., Weitzel, J. N., et al. (2009). Beyond Li-Fraumeni syndrome: clinical characteristics of families with p53 germline mutations.Journal of Clinical Oncology, 27(8), 1250–1256.
[18] Mai, P. L., Khincha, P. P., Loud, J. T., et al. (2016). Clinical management and family planning in TP53 mutation carriers: experience from the NCI Li-Fraumeni syndrome cohort.Cancer, 122(23), 3673–3681.