
{kind=link}

Cowden Syndrome Key Cowden syndrome is an autosomal dominant genetic condition characterized by benign growths called hamartomas and an increased risk of various cancers. Another name for the disease is multiple hamartoma syndrome. Llyod and Dennis described this Genodermatosis in 1963.
It is a rare disorder, and its primary cause is mutations in the phosphatase and tensin homolog gene (PTEN). This gene is located on chromosome 10q23.31. Patients with this syndrome develop macrocephaly and mucocutaneous lesions. The majority of patients develop a malignant neoplasm of the thyroid, breast, or endometrium. Management of this condition requires a collaboration of a multidisciplinary team. The prevalence of Cowden disease is approximately 1 in 200,000, but recent studies suggest it may be significantly higher due to underdiagnosis. Males and females inherit the mutated gene in equal numbers. However, some studies show a female predominance.
Etiology of Cowden Syndrome: Cowden Syndrome Key
Certain genetic mutations that you inherit are the main cause of Cowden syndrome. In this syndrome, the abnormal cells do not die as they normally do and grow uncontrollably. Excessive growth forms non-cancerous tumors or hamartomas. Researchers are still learning about the inherited gene changes that increase the risk of cancer. Mutations in the PTEN gene are considered the main cause of the condition. However, some people with this syndrome do not have a mutated gene. The other mutated genes found in the set of people suffering from Cowden syndrome are:
- SDHB-D
- PIK3CA/AKT1
- SEC23B
- KLLN
Pathophysiology of Cowden Syndrome
The PTEN is a tumour suppressor gene, as it helps control cell growth, division, and survival to prevent cells from growing uncontrollably. In Cowden syndrome, a mutation in the PTEN gene causes the loss of its normal function, leading to excessive activation of growth signalling, which allows cells to grow and divide abnormally. Hence, many benign growths (hamartomas) develop in various tissues of the body, which increases the risk of developing certain types of cancer. The disease follows an autosomal dominant inheritance pattern. A person only needs to inherit one mutated copy of the PTEN gene from a parent to have the syndrome. As not everyone with a PTEN mutation develops cancer, some additional mutations can also trigger the formation of tumors.

Illustration of common malignant pathologies associated with Cowden Syndrome. Image Courtesy: PTEN Hamartoma Tumor Syndrome/Cowden Syndrome: Genomics, Oncogenesis, and Imaging Review for Associated Lesions and Malignancy by Dragoo et al. 2021,doi.org/10.3390/cancers13133120, available via: https://www.mdpi.com/2072-6694/13/13/3120, CC BY 4.0.
Clinical features of Cowden Syndrome
The symptoms of Cowden syndrome involve both mucocutaneous and non-cutaneous features.
Mucocutaneous Features:
Mucocutaneous symptoms of this syndrome involve the following:
Hamartomas
They are the most obvious and common signs of Cowden syndrome. They are non-cancerous but tumor-like growths that can grow anywhere in your body. Hamartomas typically appear on the face, head, and neck.
Sclerotic Fibromas
They are the skin-colored, well-circumscribed, and smooth papules.
Trichilemmomas
Trichilemmomas are yellow-brown or skin colored flat, warty papules on the central face surrounding the eyes, mouth, and nose.
Oral Papillomata
They are benign wart-like epithelial tumors present particularly on the tongue, lips, gingivae, and buccal mucosa in nearly all patients of Cowden syndrome. They give a cobblestone appearance to the mucosa when present in extensive amounts.
Acral Keratoses
They are usually clear and translucent with a central indentation. These hyperkeratotic warty lesions are found on the dorsal or palmoplantar surfaces of the hands or feet. They can also appear on the trunk and face after some time.
Soft Tissue Lipomas
These are common, harmless, non-cancerous tumors. They are made up of fat cells that grow just under the skin. They often present as round, soft, and movable lumps.
Angiolipomas
Angiolipomas are benign and painful lumps beneath the skin. They often appear in multiple nodules on the arms and trunk of young adults.
Melanoma
A type of skin cancer that starts in the melanocytes.
Penile Melanosis or Lentiginosis
They are benign, asymptomatic, harmless black or brown spots on the head of the penis that are a form of hyperpigmentation.
Glycogenic Acanthosis
It is a benign, asymptomatic esophageal condition. The condition is characterized by small, white, and raised plaques of thickened squamous epithelium containing glycogen-rich cells.
Mucocutaneous Neuromas
They are benign tumors that form around the nerves in the tongue, lips, eyelids, lining of the mouth, and gastrointestinal tract.
Other Non-Cutaneous Symptoms:
Non-cutaneous features involving the central nervous system include:
- Lhermitte-Duclos disease (cerebellar dysplastic gangliocytoma). It is a benign tumor of the cerebellum and is specific to Cowden syndrome.
- Other symptoms include autism spectrum disorders, such as intellectual disability and developmental delays.
Skeletal manifestations include:
- Scoliosis
- High arched palate
- Macrocephaly
Females with Cowden syndrome can develop breast carcinoma (estimates vary from 25% to 85%). Breast cancer can also be observed in males.
Thyroid abnormalities include:
- Thyroid cancer (in almost 30% patients)
- Goiter
- Thyroid adenoma
- Thyroglossal duct cyst
Genitourinary abnormalities include:
- Endometrial cancer
- Renal cell carcinoma
Gastrointestinal anomalies include:
- Colon cancer
- Hamartomatous gastrointestinal polyps.
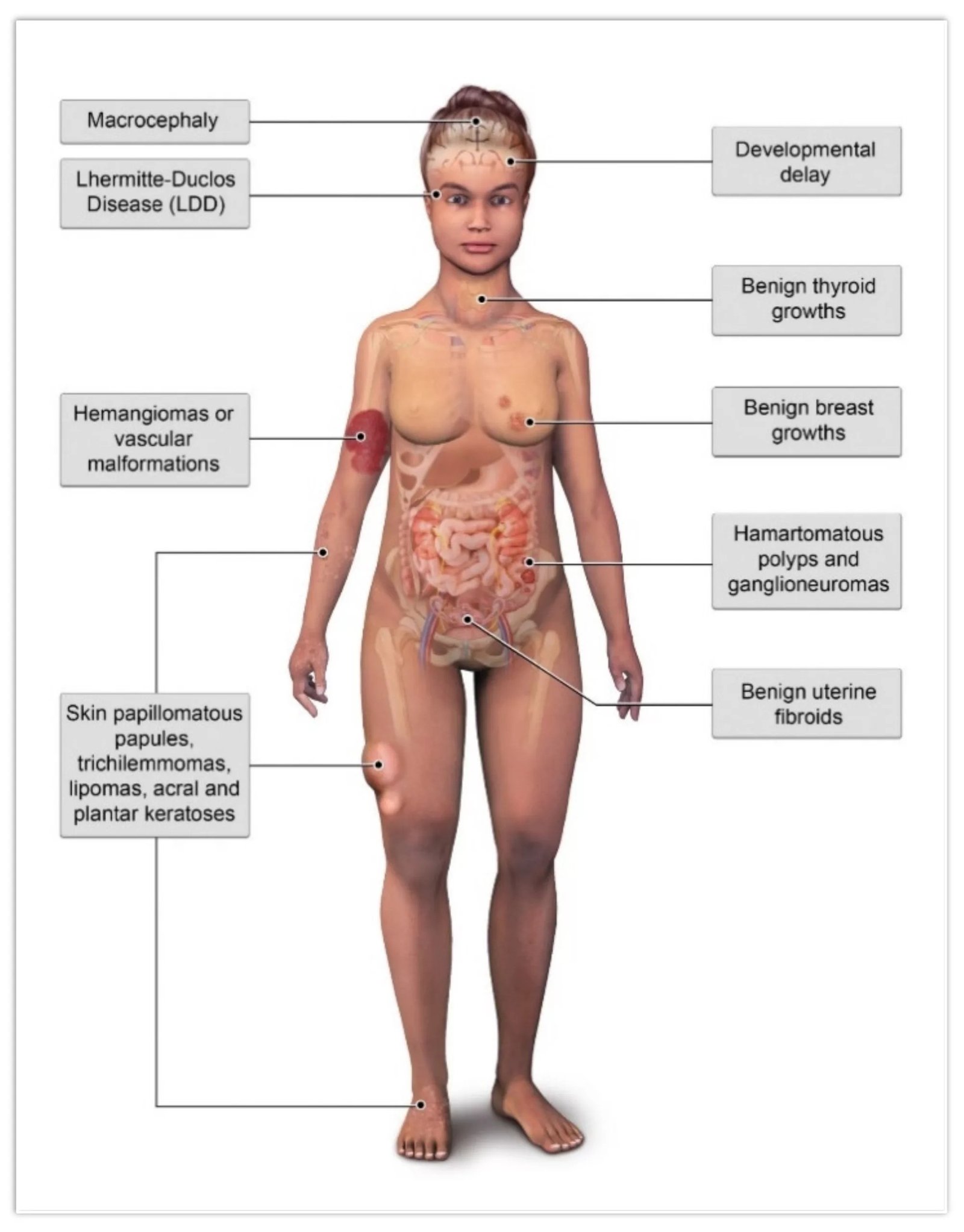
Illustration of common benign pathologies associated with Cowden syndrome.Image Courtesy: PTEN Hamartoma Tumor Syndrome/Cowden Syndrome: Genomics, Oncogenesis, and Imaging Review for Associated Lesions and Malignancy by Dragoo et al. 2021,doi.org/10.3390/cancers13133120, available via: https://www.mdpi.com/2072-6694/13/13/3120, CC BY 4.0.
Diagnosis of Cowden Syndrome
The diagnosis of Cowden syndrome relies on a combination of clinical evaluation, family history, and genetic testing. Because the syndrome can mimic other conditions and its manifestations vary widely, accurate diagnosis is often challenging. Clinicians may perform a skin biopsy when cutaneous lesions are present, but the cornerstone of diagnosis is applying established clinical criteria and confirming PTEN gene mutations. Additional investigations, such as complete blood count, stool for occult blood, thyroid function tests, urinalysis, and imaging (ultrasound, MRI, or CT), help identify associated malignancies or structural abnormalities.
The International Cowden Syndrome Consortium and later the National Comprehensive Cancer Network (NCCN) have established diagnostic criteria that guide clinicians in recognizing this condition.
History & Physical Examination:
A baseline full physical examination helps detect early changes resulting from malignancies and is an essential component of Cowden syndrome. Most patients initially present with cutaneous or mucocutaneous lesions, which often prompt referral to dermatologists. Physicians also carefully assess for macrocephaly, trichilemmomas, or oral papillomas.
A detailed family history is essential, with specific focus on relatives with thyroid, breast, or endometrial cancers, as well as gastrointestinal hamartomas, developmental abnormalities, or urological lesions. This information helps identify potential hereditary patterns and guides further testing.
Diagnostic Criteria:
The diagnostic criteria for Cowden syndrome are divided into major and minor categories. Clinical diagnosis is typically made if a patient meets either:
- Three major criteria (with one being macrocephaly, Lhermitte-Duclos disease, or gastrointestinal hamartomas), or
- Two major and three minor criteria. Confirmation with genetic testing (PTEN mutation analysis) strengthens the diagnosis.
Major Criteria
- Breast cancer
- Thyroid cancer (especially follicular type)
- Endometrial cancer
- Gastrointestinal hamartomas (including ganglioneuromas)
- Lhermitte-Duclos disease (dysplastic cerebellar gangliocytoma)
- Macrocephaly
- Multiple mucocutaneous lesions (trichilemmomas, oral papillomas, acral keratoses)
Minor Criteria
- Autism spectrum disorder
- Colon cancer
- Esophageal glycogenic acanthosis (≥3 lesions)
- Lipomas
- Intellectual disability (IQ ≤75)
- Renal cell carcinoma
- Testicular lipomatosis
- Thyroid structural lesions (goiter, adenomas, nodules)
- Vascular anomalies (hemangiomas, arteriovenous malformations)
Management & Treatment of Cowden Syndrome
Management of patients with this syndrome is interprofessional and primarily stems from patient-specific findings. The healthcare provider refers patients to appropriate specialists such as endocrinologists, gynecologists, dermatologists, gastroenterologists, neurologists, and radiologists, depending on their clinical findings.
Genetic counseling is highly recommended for affected individuals and at-risk family members.
Treatment options for cutaneous features are:
- Oral retinoids (e.g., acitretin): May temporarily reduce skin lesions, but recurrence after discontinuation is common.
- Topical 5-fluorouracil: Useful for localized lesions.
- Surgical procedures: Such as shave excisions, chemical peels, or laser resurfacing for facial papules.
- mTOR inhibitors: Currently under investigation for potential therapeutic benefits.
For cancer surveillance, patients typically undergo:
- Annual thyroid ultrasound
- Regular breast imaging (MRI or mammography starting earlier than the general population)
- Endometrial cancer screening in women
- Colonoscopy and renal imaging based on individual risk
Differential Diagnosis
Differential diagnosis of Cowden syndrome includes:
- Birt-Hogg-Dubé syndrome (facial fibrofolliculomas, renal tumors)
- Tuberous sclerosis complex or MEN1 (angiofibromas)
- Heck disease (focal epithelial hyperplasia in the oral cavity)
- Multiple endocrine neoplasia types 1 and 2B
- Bannayan-Riley-Ruvalcaba syndrome (shares PTEN mutations, but distinguished by lipomas, hemangiomas, and pigmented macules on the penis)
MacrocephalayFinal Remarks
Cowden syndrome is a rare inherited disease that can be difficult to diagnose, and you need a diagnostic professional for confirmation. Health care providers tailor treatment according to the symptoms and your situation. You may need several kinds of tests to rule out other conditions.
References
[1] Garofola, C., Z. Jamal, and G.P. Gross,Cowden disease.2018.
[2] Garofola, C., Z. Jamal, and G.P. Gross,Cowden disease.2018.
[3] Gammon, A., K. Jasperson, and M. Champine,Genetic basis of Cowden syndrome and its implications for clinical practice and risk management.The application of clinical genetics, 2016: p. 83-92.
[4] Jornayvaz, F. and J. Philippe,Mucocutaneous papillomatous papules in Cowden’s syndrome.Clinical and experimental dermatology, 2008.33(2): p. 151-153.
[5] Kieselova, K., et al.,Multiple sclerotic fibromas of the skin: an important clue for the diagnosis of Cowden syndrome.Case Reports, 2017.2017: p. bcr-2017-221695.
[6] Khandpur, U., et al.,Bilateral recurrent dysplastic cerebellar gangliocytoma (Lhermitte-Duclos disease) in Cowden syndrome: a case report and literature review.World neurosurgery, 2019.127: p. 319-325.
[7] Fackenthal, J.D., et al.,Male breast cancer in Cowden syndrome patients with germline PTEN mutations.Journal of Medical Genetics, 2001.38(3): p. 159-164.
[8] Pilarski, R., Burt, R., Kohlman, W., Pho, L., Shannon, K. M., & Swisher, E. (2013).Cowden syndrome and the PTEN hamartoma tumor syndrome: Systematic review and revised diagnostic criteria.Journal of the National Cancer Institute, 105(21), 1607–1616. https://doi.org/10.1093/jnci/djt208
[9] Pilarski, R., et al.,Cowden syndrome and the PTEN hamartoma tumor syndrome: systematic review and revised diagnostic criteria.Journal of the National Cancer Institute, 2013.105(21): p. 1607-1616.
[10] Mester, J. and C. Eng,Cowden syndrome: Recognizing and managing a not‐so‐rare hereditary cancer syndrome.Journal of surgical oncology, 2015.111(1): p. 125-130.
[11] Iacobas, I., et al.,Oral rapamycin in the treatment of patients with hamartoma syndromes and PTEN mutation.Pediatric blood & cancer, 2011.57(2): p. 321-323.