
{kind=link}

Leiomyosarcoma Malignancy Smooth Leiomyosarcoma is an aggressive tumor of smooth muscle origin.[1] Smooth muscles are present in hollow organs of the body. This tumor primarily occurs in the retroperitoneum and uterus. Occasionally, LMS can arise in the deep soft tissues of the trunk and extremities, or in blood vessel walls, due to the presence of mesenchymal tissue. The clinical presentation varies depending on the site of origin, and hence, a wide range of symptoms may be associated with it. To diagnose, image-guided core needle biopsy is the main method. Treatment is nuanced and depends upon several factors such as the age of the patient, the general physical condition, and the severity of the disease. It primarily involves surgical resection, with chemotherapy and radiotherapy being used as supportive therapy.
How Common is this?: Leiomyosarcoma Malignancy Smooth
Leiomyosarcoma accounts for 10-20% of all sarcomas.[2]The most common site for its occurrence is the retroperitoneum (the area behind the peritoneum), whereas less commonly it also affects the uterus, trunk, and extremities. Its incidence increases with increasing age. However, uterine leiomyosarcoma is an exception that mostly occurs in perimenopausal women. Out of all uterine malignancies, leiomyosarcomas comprise about 3-7%. Retroperitoneal leiomyosarcomas and the ones arising from visceral blood vessels are more common in women, whereas others are more common in men.
Cause of Leiomyosarcoma:
The exact cause of leiomyosarcoma remains unclear, as it is a mixture of genetic predisposition and somatic mutations in DNA later in life, such as RB1 gene alterations and, less commonly, PTEN mutations. gene alterations in approximately 50–70% of patients. In addition, some other conditions are also linked to leiomyosarcoma, such as:
Leiomyosarcoma Symptoms:
Being a tumor of smooth muscle, Leiomyosarcoma may not have any symptoms at first. As the tumor grows, it starts to compress surrounding structures, and hence, related symptoms appear. In some locations, such as the retroperitoneum, tumors may become excessively large before manifesting any symptoms. General symptoms include:
- Weight loss
- Nausea and Vomiting
- Pain at the tumor site
- A feeling of mass
- Fatigue or a persistent weakness
Other symptoms correspond to the system affected. Such as, in case of retroperitoneal involvement, it may cause,
- Abdominal pain
- Constipation and feeling of fullness
- Black stool
- Loss of Appetite
Whereas in the uterus, the leiomyosarcoma may cause:
- Vaginal discharge
- Pressure symptoms, such as frequent urination
- Irregular bleeding etc
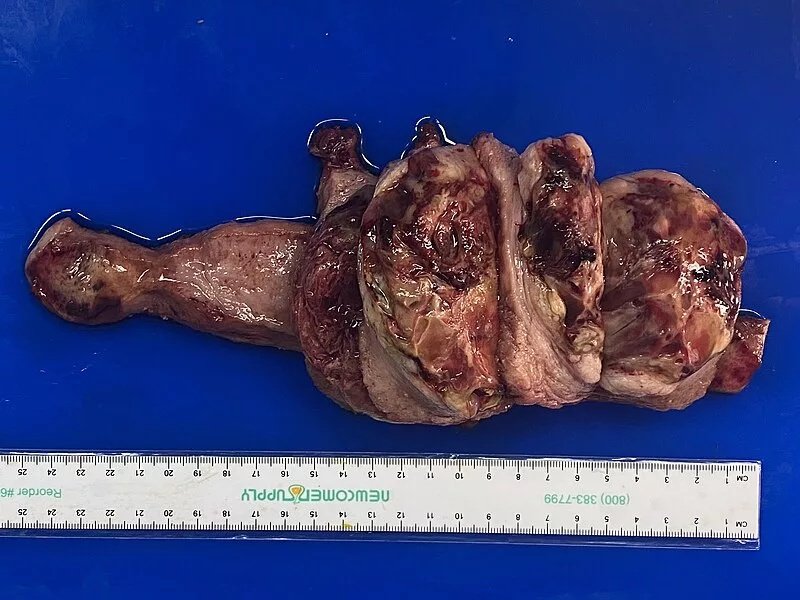
Gross pathology of uterine leiomyosarcoma showing hemorrhagic, necrotic, and cystic areas upon sectioning. Image by Mikael Häggström, M.D., viaWikimedia Commons. Licensed under CC0 (Public Domain).
Leiomyosarcoma Diagnosis:
In case of symptoms, a healthcare provider starts by taking a thorough history, which includes past medical history as well. Following it, they will do a complete physical exam, which includes palpation of the mass and looking for other cancer-related signs. After that, a series of tests is performed, such as:
1. Imaging studies:
The most common modality of initial investigation is imaging tests. These include:
Ultrasonography:
It is the very first investigation to perform in case of a suspected visceral mass. On a transvaginal ultrasound, a uterine leiomyosarcoma generally appears as a cystic mass with a necrotic center and looks like a leiomyoma. Most often, uterine leiomyosarcomas are diagnosed on biopsy after hysterectomy for leiomyomas.[4]
Computed Tomography (CT) Scan:
A CT machine is a special X-ray machine that takes many images with different cross-sections and combines them. It is the best modality to assess retroperitoneal and uterine leiomyosarcomas. Moreover, it is excellent for detecting calcification and lung metastasis.
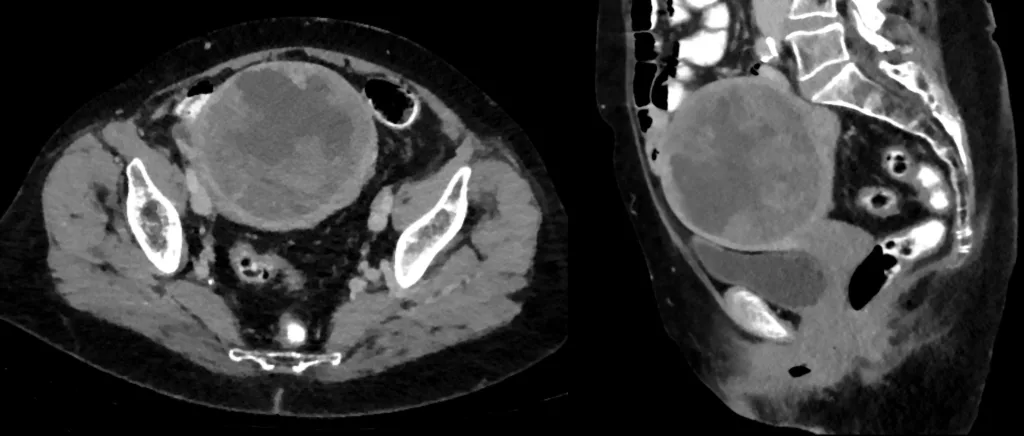
CT scan (axial and sagittal view) showing uterine leiomyosarcoma. Image by Hellerhoff, CC BY-SA 3.0, via Wikimedia Commons.
Magnetic Resonance Imaging (MRI):
MRI uses electromagnetic waves, and it is a gold standard investigation for evaluating tumours originating in the trunk and extremities.[5]Both MRI and CT scans help to identify the tumor extent, its relationship to surrounding structures, and targets for biopsy.
PET Scan:
In case of advanced sarcoma with metastasis, a PET scan may provide additional details. Moreover, it helps to distinguish scar tissue from viable tumors. The role of the PET scan is limited to clinical trials and advanced-stage disease.
2. Biopsy:
If imaging tests are suggestive of sarcoma, the next step is to take a biopsy. Usually, it includes taking samples from various parts of the cancer, and later, the pathologist observes them in the lab. A fine needle aspiration is insufficient for establishing a diagnosis. Hence, an image-guided core needle biopsy is the most accurate way.[6] It is important to be careful during the procedure on abdominal or pelvic lesions to avoid peritoneal seeding and possible sarcomatosis.
3. Other tests:
Physicians also advise baseline tests such as Complete Blood Count (CBC), Complete Urine Examination (CUE), and basic imaging such as Chest X-ray (CXR) in case of a suspected mass. While these tests may indicate overall health conditions, they don’t have any significant role in diagnosing leiomyosarcoma. Leiomyosarcoma is classified under ICD-10 (e.g., C49.0–C49.9 for soft tissue sites and C54.2 for uterine leiomyosarcoma), which helps in accurate medical documentation and tracking.
Leiomyosarcoma Treatment:
The management of leiomyosarcoma depends on the disease site, tumor size of and grade of tumor. A multi-disciplinary approach in a large centre is highly recommended to achieve better results. The primary goal of treatment includes having complete surgical resection with tumor-free margins, reducing recurrence, and preventing metastasis. Treatment options include surgery, chemotherapy, and radiotherapy.
1. Surgery (Mainstay of treatment):
In case of accessible areas where surgery is possible, it is the main treatment option. The goal of surgery is to achieve R0 resection, which means complete removal of the tumor with no microscopic cancer cells left at the margins, while preserving important neurovascular structures.
Extremity/Trunk Tumors:
In tumors of the extremities or trunk, limb-sparing surgery is preferred whenever possible. In cases where achieving negative margins (R0) would result in loss of limb function, an R1 resection (microscopic residual disease) may be accepted, provided that adjuvant radiotherapy is given to enhance local control.[7]
Retroperitoneal Tumors:
Sarcomas of the retroperitoneum often present as large tumors, and attaining negative margins is quite challenging. An en bloc resection that includes the tumor and any involved adjacent organs (such as the kidney, colon, or pancreas) offers the best chance of local control.[8] However, tumor rupture and spillage are quite common and need a careful approach. Pre-op radiation is generally ineffective.
Uterine Tumors:
Total Hysterectomy and en bloc removal are the standard surgical procedures for all uterine leiomyosarcomas. In most cases, bilateral salpingo-oophorectomy is also performed, except in selected premenopausal women, where ovarian preservation may be considered.[9] Lymph node dissection is not mandatory. In cases of extra-uterine spread, surgical resection of all visible disease should be attempted when feasible.
Metastatic/Recurrent Disease:
The National Comprehensive Cancer Network (NCCN) recommends pulmonary metastasectomy in patients where only lung metastasis is present.[10] In cases of local recurrence, surgical re-excision remains an option when technically feasible.
2. Radiotherapy:
Perioperative radiotherapy is the gold standard of treatment in localised lesions of the extremities, trunk, and head and neck region. Studies demonstrate that external beam radiotherapy and postoperative brachytherapy improve local control in sarcomas of the head and neck. The timing of Radiotherapy is still in debate. Preoperative radiotherapy has the benefit of delivering a lower total dose with a short duration of treatment. However, it causes more wound complications and slower healing. On the other hand, postoperative radiotherapy shows fewer wound complications but is associated with more fibrosis/edema.[11] In retroperitoneal and uterine sarcomas, the role of radiotherapy is limited.
3. Chemotherapy:
Leiomyosarcoma is moderately sensitive to chemotherapy.[12] Chemotherapy is of two types.
Adjuvant Chemotherapy (After Surgery):
Guidelines suggest that Adjuvant chemotherapy for tumors larger than 5cm or high-grade tumors can be a viable option. Doxorubicin is the main drug for this purpose and has better outcomes when combined with ifosfamide. Trials have shown better results in the paediatric age group than in adults.[13]
Neoadjuvant Chemotherapy (Before Surgery)
When given before surgery, chemotherapy may downstage the tumor, improve margins, and help to control the disease. In addition, it can provide essential clues in the responsiveness of the disease to chemotherapy. In some trials, Epirubicin and ifosfamide given in combination have demonstrated good outcomes in terms of achieving negative margins.
Metastatic/unresectable disease:
For advanced or unresectable disease, anthracyclines (e.g., doxorubicin) are the first-line agents. Although they rarely improve overall survival, they provide symptom relief and longer progression-free survival. Other active agents include gemcitabine, trabectedin, pazopanib, and eribulin, which may be used in second-line or refractory cases depending on patient tolerance and prior treatment.[14]
4. Immunotherapy:
Except for a few cases, Immunotherapy has not shown any promising results. Nivolumab and Ipilimumab show some responses in leiomyosarcoma and are currently in use at a limited level.[15]
Leiomyosarcoma Staging:
The staging of this cancer is according to the site of origin. Uterine leiomyosarcoma staging follows the Federation of Obstetrics and Gynaecology (FIGO)classification. The sarcomas of the head, neck, and retroperitoneum are staged by the American Joint Committee on Cancer (AJCC) staging system.
1. Tumor Grading by French Federation of Cancers (FNCLCC) System:
To grade all types of soft tissue sarcomas, AJCC recommends this 3-tiered grading system devised by the French Federation of Cancers.
In this system, grading is based on 3 factors:
- Differentiation (How much the tumor resembles normal tissues)
- Mitotic count (Number of dividing cells)
- Tumor necrosis (Amount of dead tumor tissue)
| Factor | Score Description |
|---|---|
| Differentiation | 1: Looks like normal tissue2: Unclear type3: Undifferentiated or high-grade (e.g.,Ewing, synovial sarcoma) |
| Mitotic count | 1: 0–9 mitoses per 10 HPFs2: 10–19 per 10 HPFs3: 20+ per 10 HPFs |
| Necrosis | 0: None1: <50% necrosis2: ≥50% necrosis |
Final Grade:
In the end, it entails assigning a cumulative score to the sarcoma based on all the evaluations.
- G1 (Low grade): 2–3 points
- G2 (Intermediate): 4–5 points
- G3 (High grade): 6–8 points
- GX: Grade cannot be assessed
2. AJCC 8th Edition Staging for Sarcomas of Trunk and Extremities:
TNM staging of AJCC categorises tumors on the basis of size, nodal involvement, and metastasis.
| Tumor Size (T) | T1: ≤5 cmT2: >5 cm to ≤10 cmT3: >10 cm to ≤15 cmT4: >15 cm |
|---|---|
| Lymph Nodes (N) | N0: No lymph node spreadN1: Regional lymph node involvement |
| Metastasis (M) | M0: No distant spreadM1: Distant metastasis (e.g., lungs, liver) |
Stage groups:
| Stage I | Stage IA: T1; N0; M0; G1Stage IB: T2, T3, T4; N0; M0; G1 |
|---|---|
| Stage II | T1; N0; M0; G2/3 |
| Stage III | Stage IIIA: T2; N0; M0; G2/3Stage IIIB: T3, T4; N0; M0; G2/3 |
| Stage IV | Any T; N1; M0; any G Any T; any N; M1; any G |
3. AJCC 8th edition Staging for Retroperitoneal Sarcomas:
The staging for retroperitoneal sarcomas is similar to trunk and extremities. The only difference is that patients with N1 involvement are classified under stage IIIb rather than stage IV.
4. AJCC 8th edition Staging for Head and Neck Sarcomas:
There are no formal staging groups in head and neck sarcomas in the AJCC 8th edition, as it would require a French grade system, and very extensive lesions (T4) remain unclassified. However, these sarcomas are categorized as:
| T Category | T1: ≤ 2 cmT2: > 2 cm and ≤ 4 cmT3: > 4 cmT4: Invasion of nearby structuresT4a: Invasion of the orbit, skull base, dura, central viscera, pterygoid muscles, or facial skeletonT4b: Invasion of the brain, CNS via perineural spread, prevertebral muscle, or carotid artery |
|---|---|
| N Category | N0: No lymph node involvementN1: Regional lymph node involvement |
| M Category | M0: No distant metastasisM1: Distant metastasis |
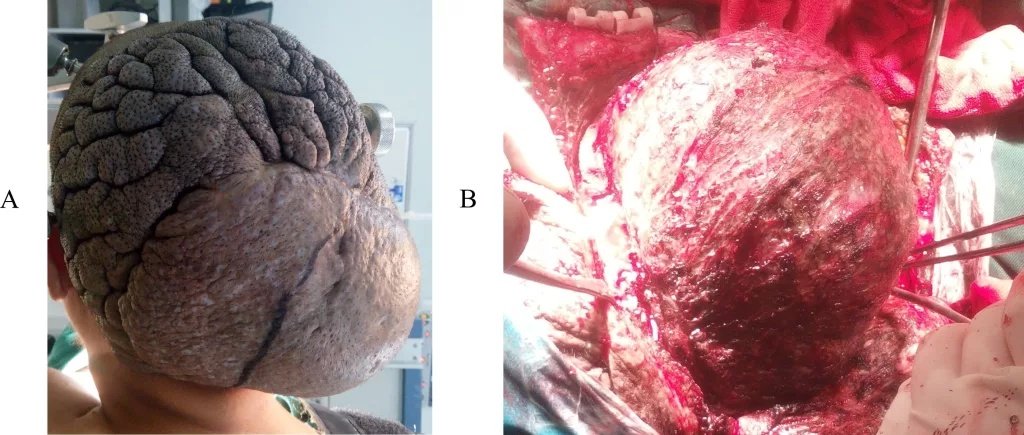
Leiomyosarcoma of the scalp. (A) Localized cutis verticis gyrata with a 12 cm hard, painless mass in the occipital and vertex region; the lesion was enlarging. (B) The tumor was exposed during surgical resection.Image by Gau, Liu, Liu, Yang, and Yang, published in Frontier, licensed under CC-BY.
5. FIGO Staging for Uterine Leiomyosacomas (2009):
| Stage II: Tumor extends beyond the uterus, within pelvis | IA: < 5 cm in greatest dimensionIB: > 5 cm in greatest dimension |
|---|---|
| Stage II: Tumor extends beyond the uterus, within the pelvis | IIA: Adnexal involvementIIB: Other pelvic tissues involvement |
| Stage III: Tumor invades abdominal tissues | IIIA: One siteIIIB: More than one siteIIIC: Involves pelvic and/or para-aortic lymph nodes |
| Stage IV: Tumor invades pelvic organs or distant sites | IVA: Involvement of bladder or rectumIVB: Distant metastases |
Differential Diagnoses:
Since the clinical presentation of the disease is vague and non-specific, many other conditions can present with similar symptoms or histopathological features. Hence, differentiating on the basis of microscopic examination, immunohistochemistry, and molecular testing is crucial. The conditions that mimic the disease include:
- Meningioma
- Gastrointestinal stromal tumor
- Leiomyoma
- Liposarcoma
- Endometrial stromal sarcoma
- Inflammatory myofibroblastic tumor
- Perivascular epithelioid cell tumor
Leiomyosarcoma Prognosis:
The prognosis of leiomyosarcoma depends on multiple factors such as histologic grade, tumor size, and tumor depth. Out of them, histologic grade remains the strongest predictor. The reason behind this is that most leiomyosarcomas are high-grade (grade 2–3) and therefore carry a high risk of distant metastasis and reduced survival.[16] Similarly, tumors larger than 10 cm, those involving bone or neurovascular structures, and those located deep within tissues further worsen the prognosis. Tumor location also plays a crucial role. Extremity tumors show better outcomes than retroperitoneal or visceral ones.
Moreover, Memorial Sloan Kettering has developed a nomogram, which incorporates multiple variables to assess the 5-year survival rate. The monogram performed better in predicting survival rate than the traditional FIGO or AJCC systems. Finally, in metastatic disease, younger patients with good performance status and longer disease-free intervals achieve better outcomes, especially when they receive combination chemotherapy such as doxorubicin and ifosfamide.
Why Is Leiomyosarcoma So Deadly?
Leiomyosarcoma is considered deadly due to its aggressive nature, high likelihood of metastasis, and often late diagnosis. High-grade tumors, deep location within tissues, or large tumor size further increase the risk of recurrence and reduce overall survival, making early detection and complete surgical resection crucial.
Conclusion:
Leiomyosarcoma is a rare but aggressive malignant tumor of smooth muscle origin. It often shows poor outcomes due to its high metastatic potential and late diagnosis. However, early detection, accurate histopathological assessment, and multidisciplinary management lead to a better overall survival rate. Surgical resection with clear margins remains the cornerstone of treatment. Additionally, adjuvant therapies are also in use based on tumor grade and stage. Ongoing research into molecular pathways and targeted therapies offers hope for better prognostic tools and treatment strategies in the future. Recurrence and metastasis can occur even years after initial treatment, making regular follow-up crucial.
References
[1] Martin-Liberal J. Leiomyosarcoma: Principles of management. Intractable Rare Dis Res. 2013 Nov;2(4):127-9.
[2] Devaud N, Vornicova O, Abdul Razak AR, Khalili K, Demicco EG, Mitric C, Bernardini MQ, Gladdy RA. Leiomyosarcoma: Current Clinical Management and Future Horizons. Surg Oncol Clin N Am. 2022 Jul;31(3):527-546.
[3] George S, Serrano C, Hensley ML, Ray-Coquard I. Soft Tissue and Uterine Leiomyosarcoma. J Clin Oncol. 2018 Jan 10;36(2):144-150.
[4] George S, Serrano C, Hensley ML, Ray-Coquard I. Soft Tissue and Uterine Leiomyosarcoma. J Clin Oncol. 2018 Jan 10;36(2):144-150.
[5] Demas BE, Heelan RT, Lane J, Marcove R, Hajdu S, Brennan MF. Soft-tissue sarcomas of the extremities: comparison of MR and CT in determining the extent of disease. AJR Am J Roentgenol. 1988 Mar;150(3):615-20.
[6] Strauss DC, Qureshi YA, Hayes AJ, Thway K, Fisher C, Thomas JM. The role of core needle biopsy in the diagnosis of suspected soft tissue tumours. J Surg Oncol. 2010 Oct 01;102(5):523-9.
[7] Byerly S, Chopra S, Nassif NA, Chen P, Sener SF, Eisenberg BL, Tseng WW. The role of margins in extremity soft tissue sarcoma. J Surg Oncol. 2016 Mar;113(3):333-8.
[8] Bonvalot S, Miceli R, Berselli M, Causeret S, Colombo C, Mariani L, Bouzaiene H, Le Péchoux C, Casali PG, Le Cesne A, Fiore M, Gronchi A. Aggressive surgery in retroperitoneal soft tissue sarcoma carried out at high-volume centres is safe and is associated with improved local control. Ann Surg Oncol. 2010 Jun;17(6):1507-14.
[9] Seagle BL, Sobecki-Rausch J, Strohl AE, Shilpi A, Grace A, Shahabi S. Prognosis and treatment of uterine leiomyosarcoma: A National Cancer Database study. Gynecol Oncol. 2017 Apr;145(1):61-70.
[10] Von Mehren M, Kane JM, Agulnik M, Bui MM, Carr-Ascher J, Choy E, Connelly M, Dry S, Ganjoo KN, Gonzalez RJ, Holder A, Homsi J, Keedy V, Kelly CM, Kim E, Liebner D, McCarter M, McGarry SV, Mesko NW, Meyer C, Pappo AS, Parkes AM, Petersen IA, Pollack SM, Poppe M, Riedel RF, Schuetze S, Shabason J, Sicklick JK, Spraker MB, Zimel M, Hang LE, Sundar H, Bergman MA. Soft Tissue Sarcoma, Version 2.2022, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2022 Jul;20(7):815-833.
[11] Tiong SS, Dickie C, Haas RL, O’Sullivan B. The role of radiotherapy in the management of localized soft tissue sarcomas. Cancer Biol Med. 2016 Sep;13(3):373-383.
[12] Grimer R, Judson I, Peake D, Seddon B. Guidelines for the management of soft tissue sarcomas. Sarcoma. 2010;2010:506182.
[13] udson, I., Verweij, J., Gelderblom, H., Hartmann, J. T., Schöffski, P., Blay, J. Y., Kerst, J. M., Sufliarsky, J., Whelan, J., & Hohenberger, P. (2014).Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: A randomised controlled phase 3 trial.The Lancet Oncology, 15(4), 415–423. https://doi.org/10.1016/S1470-2045(14)70063-4
[14] Van der Graaf, W. T. A., Blay, J. Y., Chawla, S. P., Kim, D. W., Bui-Nguyen, B., Casali, P. G., Schöffski, P., Aglietta, M., Staddon, A. P., & Beppu, Y. (2012).Pazopanib for metastatic soft-tissue sarcoma (PALETTE): A randomised, double-blind, placebo-controlled phase 3 trial.The Lancet, 379(9829), 1879–1886. https://doi.org/10.1016/S0140-6736(12)60651-5
[15] D’Angelo, S. P., Mahoney, M. R., Van Tine, B. A., Atkins, J., Milhem, M. M., Horvath, E. L., Tap, W. D., Schwartz, G. K., Streicher, H., & Conley, A. P. (2018).Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): Two open-label, non-comparative, randomised, phase 2 trials.The Lancet Oncology, 19(3), 416–426. https://doi.org/10.1016/S1470-2045(18)30006-8
[16] Pisters PW, Leung DH, Woodruff J, Shi W, Brennan MF. Analysis of prognostic factors in 1,041 patients with localized soft tissue sarcomas of the extremities. J Clin Oncol. 1996 May;14(5):1679-89.