
 is a rare and highly malignant brain cancer of the pineal gland, a small structure in the brain...){kind=link}

Pineoblastoma This Rare Pineoblastoma (PB) is a rare and highly malignant brain cancer of the pineal gland, a small structure in the brain. Genetic mutations cause pineal cells to lose control over their normal process of division, which leads them to multiply without limits and form a tumor. These cancerous cells enter the cerebrospinal fluid (CSF), where they travel and seed secondary tumors throughout the whole central nervous system (CNS). Early diagnosis and intensive treatment of PB are therefore critical for survival.
What is the Pineal Gland?: Pineoblastoma This Rare
Our pineal gland is a small pine-shaped endocrine organ buried deep in our brain that releases the melatonin hormone, which helps our body regulate the sleep-wake cycle.
The pineal gland also plays antioxidant roles and modulates the reproductive hormone-releasing timing through its effect on the hypothalamic–pituitary axis.
Pathophysiology & Causes of Pineoblastoma
Genetic mutations, either inherited or acquired, are the main culprit of pineoblastoma. These mutations break the normal control over cell growth and maturation, leading to unchecked and rapid cell division.
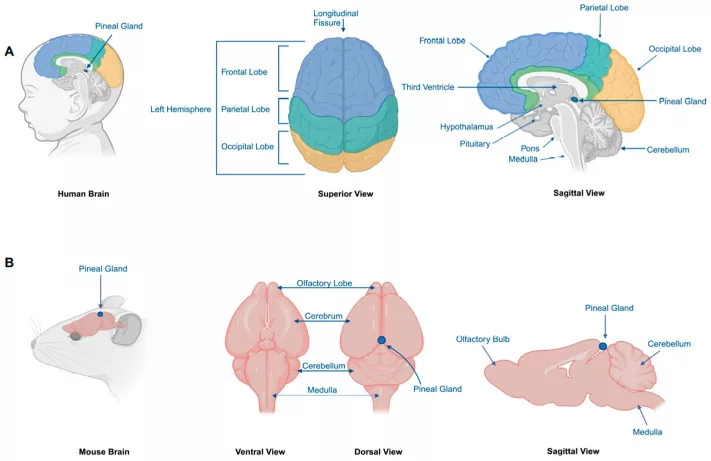
Image courtesy: “Figure 1” from Recent Advances in Pineoblastoma Research by Jiang et al., 2025, published in Cancers (MDPI), licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). Source: PubMed Central – PMC11898778
Inherited Genetic Mutations:
In some cases, inherited genetic mutations increase the risk of developing pineoblastoma.
- RB1 (Retinoblastoma) Mutation: The RB1 gene limits cell division, but when this gene is altered or lost, cells divide repeatedly and uncontrollably. In children with inherited RB1 mutations, there is an increased risk of developing PB.retinoblastoma: a systematic review and meta-analysis.The Lancet. Oncology,15(10), 1157–1167.” style=”position:relative;color:#309b65;cursor:help;border-bottom:1px dotted #309b65;font-weight:bold”>[1]
- DICER1 Syndrome: Mutations in the DICER1 gene lead to multiple cancers in the body, including pineoblastoma. This gene also has a role in the production of microRNAs, the small regulators that decide which genes should be silenced and which should be expressed. Children with the DICER1 gene alterations lose this gene-balancing activity, allowing the pineal cells to grow rapidly to cause pineoblastoma.[2]
- DROSHA Mutations: The DROSHA gene works together with the DICER1 gene to regulate other genes through microRNA biogenesis. Recent studies report that the DROSHA gene mutations are also linked with PB.[3]
Acquired Genetic Mutations:
Most cases of pineal gland tumors arise due to acquired genetic mutations. These are:
- MicroRNA-Processing Gene Loss: Acquired mutations in the DROSHA or DICER1 genes, similar to inherited mutations, can also cause PB.
- RB1 Loss: In many cases of pineoblastoma, the RB1 gene is lost randomly during the life of the patient, removing the checks on cell division.
- MYC Activation & FOXR2 Overexpression: The MYC gene speeds up cell division. In PB, the MYC gene is abnormally activated, along with another gene, FOXR2, resulting in an increased cell division rate.[4]
Not all the causes of PB are due to genetic mutations; there are some other causes, like abnormal DNA methylation. These methylations are chemical tags that reprogram pineal cells toward an immature state and block normal differentiation, promoting rapid growth.[5]
Metastatic Spread of Pineoblastoma:
As the PB tumor grows, it compresses nearby structures, and some metastatic cells may break off from the main mass, invade the brain coverings, and enter the CSF, from where these cancerous cells spread further.
- Growth & local invasion: Once the pineal gland tumor develops, it starts to grow rapidly, pressing the nearby structures (midbrain, thalamus, and third ventricle). It also blocks the normal flow of CSF, resulting in increased intracranial pressure, causing clinical symptoms of headache, nausea, and vomiting.
- Spread within the Brain & Spinal Cord: Cancerous cells in pineoblastoma invade the CSF, causing these cells to circulate throughout the brain and spinal cord. This spread is called leptomeningeal dissemination, by which cancer cells seed new tumors at secondary sites throughout the entire CNS.
- Systemic Metastasis: In most cases, the spread of pineoblastoma remains limited to the brain and spinal cord. Systemic metastases (bone, lung, liver) are very rare and usually occur in advanced or recurrent disease.
Stages of Pineoblastoma
Tumor staging is important to know how far the cancer has spread at the time of diagnosis, to help doctors decide the treatment plan and options, and estimate prognosis.
Primary Tumor (T Staging):
- T1: The tumor is less than 3 cm in size, limited to the pineal region, and not spreading into nearby structures.
- T2: The tumor is larger than 3 cm but still mostly limited to the pineal gland area.
- T3: The tumor has grown into the neighboring brain parts, such as the third ventricle or midbrain.
- T4: The tumor has reached even further, reaching the upper brainstem or the cervical (upper spinal) canal.
Metastatic Spread (M Staging):
Because pineoblastomas often spread through the CSF, doctors also grade the disease using “M” stages:
- M0: No evidence of spread beyond the pineal region.
- M1: Tumor cells are found floating in the CSF, but no visible tumors elsewhere.
- M2: Small deposits of tumor are found inside the brain but away from the pineal gland.
- M3: Tumor deposits are found in the spinal cord lining or spinal canal.
- M4: Rare cases where the tumor has spread outside the central nervous system to organs like bone, liver, or lungs.
For ease, many oncologists use two broad categories:
- Localized Pineoblastoma (M0): Tumor confined to the pineal region, no spread.
- Metastatic Pineoblastoma (M1–M4): Tumor has spread in the brain, spinal cord, or (rarely) body.
WHO Grading of Pineoblastoma:
WHO classifies pineoblastoma as a WHO Grade IV tumor because of its aggressive behavior and high risk of leptomeningeal spread.
How common is Pineoblastoma?
Pineoblastoma is extremely rare, far less than 1%. One large population study in the United States claimed an age-adjusted incidence rate (AAIR) of 0.049 per 100,000 for children aged 0–4.[6]
Most commonly, it is seen in very young children, with the peak incidence in children under 4 years old. Pineoblastoma is also more common in Black children compared to White children.
Symptoms of Pineoblastoma
The symptoms of pineoblastoma vary depending on the blockage of the CSF flow and pressure on the nearby brain regions.
Hydrocephalus:
The increased intracranial pressure (ICP) due to the blockage of CSF by the growing tumor results in a condition known as hydrocephalus, which clinically presents as:
- Persistent or worsening headache
- Nausea and vomiting
- Lethargy
- Drowsiness
- Changes in the mental status
Young children and infants may face:
- Irritability
- Poor feeding
- Delayed milestones
Most people with pineal-region cancers develop hydrocephalus. It is important to recognize hydrocephalus immediately to treat the patient at the initial stages of the disease.
Visual & Eye Movement Problems:
The growing pineal tumor presses on the nearby regions of the brain, such as the midbrain and tectal area, leading to Parinaud’s syndrome, a set of eye-movement abnormalities in which you may feel:
- Difficulty in looking upward (Upward gaze palsy)
- Double vision
- Retraction of eyelids
- Abnormal pupil reaction
- Convergence-retraction nystagmus is a condition of jerky backward movement of the eyes when attempting to look up.
These eye movement abnormalities are a characteristic sign of pineal-region lesions.[7]
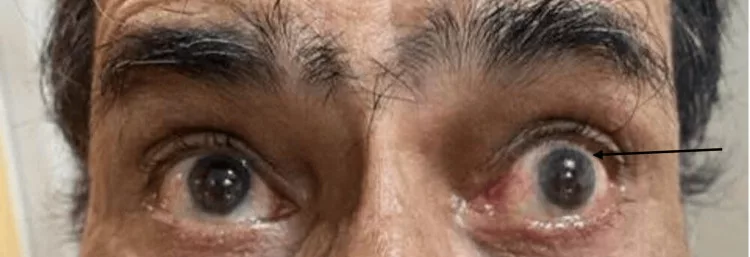
Image courtesy: Magdum et al., “Parinaud’s Syndrome: A Case Report,” Cureus, 2024 (16[4]:e58120). Licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). Source: PubMed Central (PMC)
Leptomeningeal Carcinomatosis:
Cancerous cells in pineoblastoma may spread to the CSF, causing a condition called leptomeningeal carcinomatosis, where tumor cells circulate and implant inside the whole brain and spinal cord.[8]
Common Cranial Symptoms
- Facial numbness
- Hearing problems
- Swallowing difficulties
Common Spinal Symptoms
- Backpain
- Leg weakness or numbness
- Change in gait
- Bladder or bowel dysfunction
You may have seizures when the tumor enlarges enough to infiltrate or compress the cortex of your brain. Some long-term surviving patients may experience episodes of epilepsy, which are usually related to treatment.
Diagnosis of Pineoblastoma
Pineoblastoma’s accurate and timely diagnosis is important, which involves the careful examination of physical symptoms, brain MRI, study of tumor markers, and biopsy.
- Physical Examination & History: Your physical exam focuses on neurologic signs, including eye-movement testing for Parinaud’s syndrome and a detailed family history to identify possible inherited cancer syndromes. In patients with RB1 mutations, your oncologist may recommend an ophthalmologic examination to screen for retinoblastoma, a type of eye cancer.
- Brain MRI: Pineoblastoma shows large, irregular masses on the brain MRI, highlighting the brain hemorrhage and tissue necrosis in the pineal gland region. These features, along with hydrocephalus, raise strong concerns for the PB.
- Tumor Markers: To differentiate PB from other cancers of the pineal gland, your clinician may test for blood and CSF for the specific tumor markers, the alpha-fetoprotein (AFP) and beta-human chorionic gonadotropin (β-hCG). The rise of these markers points toward the germ cell tumors.
- Entire Spine MRI: As the cancerous cells spread to the CSF, it is necessary for the doctors to thoroughly observe the whole spine through an entire spine MRI to rule out any secondary tumors.
- CSF Cytology: For high accuracy, pathologists take a CSF sample to study it under the microscope. When you have high intracranial pressure, your CSF sampling is delayed until pressure is relieved by endoscopic third ventriculostomy. Once the ICP is lowered to safe levels, your doctor may prefer lumbar CSF because it is more sensitive than ventricular/shunt CSF.[9]
- Biopsy: Neurosurgeons often combine ETV with endoscopic biopsy in the same procedure if anatomy allows, providing both CSF diversion and tissue diagnosis. The cells and tissues taken during the biopsy are then studied under the microscope to confirm the diagnosis of pineoblastoma.[10]

Surgical approaches to pineal region tumors: Endoscopic transventricular third ventriculostomy and biopsy with CSF sampling (A), interhemispheric parietal approach (B), suboccipital transtentorial approach (C), and supracerebellar infratentorial approach (D).Image courtesy: “Figure 1” from Recent Advances in Pineoblastoma Research by Jiang et al., published in Cancers (MDPI, Basel, Switzerland, 2025). Licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). Source: PMC11898778
Pineoblastoma vs Pineocytoma
Although pineoblastoma and pineocytoma are both tumors of the pineal gland, pineoblastoma is aggressive and malignant, whereas pineocytoma usually remains locally confined.
| Feature | Pineoblastoma | Pineocytoma |
|---|---|---|
| WHO Grade | Grade 4 (high-grade, malignant) | Grade 1 (low-grade, benign) |
| Typical Age | Children, especially those under 4 years old | Adults, usually 30–60 years old |
| Growth & Behavior | Very aggressive, fast-growing, spreads through CSF pathways | Slow-growing, localized, rarely spreads |
| Symptoms | Early hydrocephalus (headaches, vomiting, vision changes), rapid neurological decline | Often, incidental or slowly progressive headaches and vision problems |
| Imaging (MRI/CT) | Large, poorly defined mass with frequent calcification and necrosis, often looks like it is invading surrounding structures | Well-circumscribed, smaller mass which may show “exploded” peripheral calcifications |
| Histology (Microscope) | Small, round blue cells, high nuclear/cytoplasmic ratio, brisk mitoses, necrosis, Homer–Wright rosettes | Well-differentiated pineal parenchymal cells, lobulated pattern, low mitotic activity |
| Immunohistochemistry | Neuronal markers (synaptophysin, CRX, OTX2), very high Ki-67 | Neuronal markers positive, low Ki-67 |
| Molecular Features | Multiple subgroups (RB1, miRNA-processing defects, MYC/FOXR2-driven) | Lacks the aggressive molecular drivers as seen in pineoblastoma |
| Prognosis | Poor, 5-year survival ~40–60% even with aggressive therapy | Excellent, >90% long-term survival after surgical removal |
| Treatment | Multimodal: surgery + craniospinal irradiation + chemotherapy | Often, surgery alone, or radiotherapy only if incomplete resection |
Management & Treatment of Pineoblastoma
Once the disease is diagnosed, the next step is its treatment. Pineoblastoma is one of the most aggressive tumors that lies deep in the brain, with a tendency to spread to the CSF, making its treatment complex.
Initial Emergency Care:
Patients of PB usually come with hydrocephalus and its symptoms. To lower the elevated intracranial pressure, neurosurgeons usually perform an endoscopic third ventriculostomy (ETV). If ETV is not feasible, intracranial pressure (ICP) can be reduced using an external ventricular drain (EVD) or a ventriculoperitoneal (VP) shunt.
Surgical Management:
After managing the hydrocephalus, doctors try to remove the tumor mass as much as possible. Although gross-total resection has better outcomes, it is difficult to perform due to the complex location of the pineal gland.
Radiations:
Radiation is the main treatment option for children and people who can safely tolerate it.
- Craniospinal Irradiations (CSI): As PB spreads to the leptomeninges, your doctor uses the CSI method to treat the tiny cancerous deposits along the whole CNS. Dosing and timing must be adapted for infants, often using chemotherapy first and delaying radiation therapy.[11]
- Proton Beam Therapy (PBT): The clinicians prefer PBT in children to reduce the radiation dose and to lower the late effects of radiation. Early studies suggest that PBT has good tumor control and lowers the tissue dose; however, its long-term outcome data are limited.[12]
Radiation helps local control of the tumor with an improved survival rate when combined with surgery and chemotherapy.
Chemotherapy:
Chemotherapy is usually done before and after the radiotherapy. Cisplatin or carboplatin, vincristine, cyclophosphamide, and etoposide are the common drug agents used for the treatment of PB.
Your healthcare provider may use High-dose chemotherapy with Autologous stem cell rescue (HDC + ASCR) in selected high-risk patients and infants to intensify systemic therapy and, in some protocols, to delay or reduce craniospinal irradiation.[13]
Supportive care, Rehabilitation & Long-term Follow-up:
Pineoblastoma treatment has its own short-term (infection, nausea, hearing loss) and long-term side effects (neurocognitive impairment, hormonal deficiencies, growth failure, and secondary cancers)
- Endocrine monitoring for pituitary dysfunction (growth hormone, thyroid, adrenal, gonadal problems) after radiation or surgery.
- Neurocognitive assessment and school support: rehabilitation, individualized education plans, cognitive therapy.
- Audiology testing is necessary because platinum drugs (cisplatin) can damage hearing.
- Fertility counseling for adolescents and fertility preservation when possible before gonadotoxic therapy.
- Regular MRI surveillance of the brain (and spine as indicated) with schedules adapted by risk and institution.
Prognosis & Life Expectancy of Pineoblastoma
Prognosis depends on many factors, including age of diagnosis, metastasis, extent of resection, molecular subgroups, and treatment intensity.
- Children above 3 years old may have a longer survival rate. Five-year survival rates are reported to be significantly higher (55–70%)depending on the series and risk group.[14]
- Children under 3 years have a worse prognosis. Five-year survival rates are reported to have dropped markedly. Some studies show that only about 15% survive 5 years.[15]
- Children with a tumor that has spread usually do worse than those whose tumor is only in the pineal gland.
Conclusion
Pineoblastoma is a rare brain tumor of the Pineal gland, the tiny gland in the brain that controls the sleep-wake cycle and some reproductive functions. PB is a lethal tumor because cancerous cells spread into the CSF and seed secondary tumors throughout the entire CNS.
It mainly affects children under 4 years old due to inherited genetic mutations like RB1 mutation, DICER1 syndrome, and DROSHA mutations. As it spreads to different parts of the brain and spinal cord, the symptoms become more severe, starting from simple headaches and nausea to Parinaud’s syndrome, convergence-retraction nystagmus, and hearing and swallowing difficulties. History, MRI scans, and biopsy help doctors diagnose it.
References
[1] de Jong, M. C., Kors, W. A., de Graaf, P., Castelijns, J. A., Kivelä, T., & Moll, A. C. (2014). Trilateral retinoblastoma: a systematic review and meta-analysis.The Lancet. Oncology,15(10), 1157–1167.
[2] Sabbaghian, N., Druker, H., Weber, E., Hamel, N., Miller, S., Choong, C. S., Gottardo, N. G., Kees, U. R., Rednam, S. P., Jongmans, M. C., Jhangiani, S., Lupski, J. R., Zacharin, M., Bouron-Dal Soglio, D., Huang, A., Priest, J. R., Perry, A., Mueller, S., Albrecht, S.,… Foulkes, W. D. (2014). Germ-line and somatic DICER1 mutations in pineoblastoma.Acta Neuropathologica,128(4), 583. https://doi.org/10.1007/s00401-014-1318-7
[3] uang, Z., Ren, X., & Hu, J. (2025). Drosha: a new tumor suppressor in pineoblastoma.Genes & development,39(11-12), 677–678. https://doi.org/10.1101/gad.352932.125
[4] asiljevic A. (2023). Histopathology and molecular pathology of pediatric pineal parenchymal tumors.Child’s nervous system: ChNS: official journal of the International Society for Pediatric Neurosurgery,39(9), 2273–2284. https://doi.org/10.1007/s00381-022-05637-x
[5] Jiang, Z., Allkanjari, M. S., D Chung, P. E., Tran, H., Ghanbari-Azarnier, R., Wang, Y., Lin, D. J., Min, J. Y., Ben-David, Y., & Zacksenhaus, E. (2025). Recent Advances in Pineoblastoma Research: Molecular Classification, Modelling and Targetable Vulnerabilities.Cancers,17(5), 720. https://doi.org/10.3390/cancers17050720
[6] Greppin, K., Cioffi, G., Waite, K. A., Ostrom, Q. T., Landi, D., Takaoka, K., Kruchko, C., & Barnholtz-Sloan, J. S. (2022). Epidemiology of pineoblastoma in the United States, 2000-2017.Neuro-oncology practice,9(2), 149–157. https://doi.org/10.1093/nop/npac009
[7] Shields, M., Sinkar, S., Chan, W., & Crompton, J. (2017). Parinaud syndrome: a 25-year (1991-2016) review of 40 consecutive adult cases.Acta ophthalmologica,95(8), e792–e793. https://doi.org/10.1111/aos.13283
[8] Cocito, C., Martin, B., Giantini-Larsen, A. M., Valcarce-Aspegren, M., Souweidane, M. M., Szalontay, L., Dahmane, N., & Greenfield, J. P. (2023). Leptomeningeal dissemination in pediatric brain tumors. Neoplasia (New York, N.Y.), 39, 100898. https://doi.org/10.1016/j.neo.2023.100898 Symptoms vary depending on the parts of the brain affected.
[9] Gajjar, A., Fouladi, M., Walter, A. W., Thompson, S. J., Reardon, D. A., Merchant, T. E., Jenkins, J. J., Liu, A., Boyett, J. M., Kun, L. E., & Heideman, R. L. (1999). Comparison of lumbar and shunt cerebrospinal fluid specimens for cytologic detection of leptomeningeal disease in pediatric patients with brain tumors.Journal of clinical oncology: official journal of the American Society of Clinical Oncology,17(6), 1825–1828. https://doi.org/10.1200/JCO.1999.17.6.1825
[10] Morgenstern, P. F., Osbun, N., Schwartz, T. H., Greenfield, J. P., Tsiouris, A. J., & Souweidane, M. M. (2011). Pineal region tumors: an optimal approach for simultaneous endoscopic third ventriculostomy and biopsy.Neurosurgical focus,30(4), E3. https://doi.org/10.3171/2011.2.FOCUS10301
[11] Lombardi, G., Poliani, P. L., Manara, R., Berhouma, M., Minniti, G., Tabouret, E., Razis, E., Cerretti, G., Zagonel, V., Weller, M., & Idbaih, A. (2022). Diagnosis and Treatment of Pineal Region Tumors in Adults: A EURACAN Overview.Cancers,14(15), 3646. https://doi.org/10.3390/cancers14153646
[12] Perkins, S. M., Prime, S., Watts, M., Huang, J., & Zhao, T. (2023). Pediatric Experience and Outcomes from the First Single-Vault Compact Proton Therapy Center.Cancers,15(16), 4072. https://doi.org/10.3390/cancers15164072
[13] Panosyan, E. H., Ikeda, A. K., Chang, V. Y., Laks, D. R., Reeb, C. L., Bowles, L. V., Lasky, J. L., 3rd, & Moore, T. B. (2011). High-dose chemotherapy with autologous hematopoietic stem-cell rescue for pediatric brain tumor patients: a single institution experience from UCLA. Journal of Transplantation, 2011, 740673. https://doi.org/10.1155/2011/740673
[14] Tate, M., Sughrue, M. E., Rutkowski, M. J., Kane, A. J., Aranda, D., McClinton, L., McClinton, L., Barani, I. J., & Parsa, A. T. (2012). The long-term postsurgical prognosis of patients with pineoblastoma.Cancer,118(1), 173–179. https://doi.org/10.1002/cncr.26300
[15] Tate, M., Sughrue, M. E., Rutkowski, M. J., Kane, A. J., Aranda, D., McClinton, L., McClinton, L., Barani, I. J., & Parsa, A. T. (2012). The long-term postsurgical prognosis of patients with pineoblastoma.Cancer,118(1), 173–179. https://doi.org/10.1002/cncr.26300