
{kind=link}

Systemic Mastocytosis Symptoms Systemic Mastocytosis is a group of disorders characterized by the excessive proliferation and accumulation of pathologic mast cells in the tissues. The term “Systemic Mast Cell Disease (SMCD)” is an older name for this condition. It belongs to a group of disorders called myeloproliferative neoplasms, which are conditions where certain blood-forming cells in the bone marrow grow and multiply abnormally. In systemic mastocytosis, this involves abnormal proliferation of mast cells, a type of white blood cell that normally helps in allergic responses and inflammation.
Mastocytosis can involve skin (cutaneous mastocytosis) or extracutaneous tissues (systemic mastocytosis). Cutaneous mastocytosis has a benign presentation, while systemic mastocytosis can be an indolent or aggressive form of the disorder. It causes the release of multiple vasoactive mediators from mast cells, resulting in a wide range of symptoms.
Anaphylaxis, diarrhea, and pruritus are the most common symptoms associated with this condition. It is a very rare disorder that affects both males and females equally. The estimated prevalence is approximately 1 in 10,000–20,000 individuals, although many cases may go undiagnosed.[1] The condition cannot be completely cured, and it tends to persist chronically, but treatment can help manage symptoms and prevent complications.[2]
Variants of Systemic Mastocytosis: Systemic Mastocytosis Symptoms
There are several subtypes of systemic mastocytosis, classified according to disease severity and organ involvement. These include:
Indolent Systemic Mastocytosis (ISM):
It is the most common stage. It is a slow-growing type that does not affect organ function.
Smoldering Systemic Mastocytosis (SSM):
SSM is also a slow-growing stage. However, it involves a higher mast cell burden and early evidence of organ involvement.
Aggressive Systemic Mastocytosis (ASM):
It is an advanced stage that involves severe organ damage due to mast cell infiltration.
Aggressive systemic mastocytosis. Aspirate smears show areas with numerous hypogranular and/or spindle-shaped Mast Cells. Image Courtesy: Review and Updates on Systemic Mastocytosis and Related Entities by Li et al, 2023,doi.org/10.3390/cancers15235626, available via: https://www.mdpi.com/2072-6694/15/23/5626 CC BY 4.0.
Systemic Mastocytosis with an Associated Hematologic Neoplasm (SM-AHN):
This subtype involves systemic mastocytosis along with another hematologic malignancy, such as myelodysplastic syndrome or myeloproliferative neoplasm.
Mast Cell leukemia (MCL):
MCL is the most severe and aggressive stage of the disease. A high number of abnormal mast cells in the blood and bone marrow characterizes this condition. It can be fatal.

Atypical mast cells (Wright–Giemsa stain, 1000×). (A) Spindle-shaped (type I) mast cell, (B) bilobed promastocyte (type II; black arrow), and (C) metachromatic blasts (black arrows). Image Courtesy: Review and Updates on Systemic Mastocytosis and Related Entities by Li et al, 2023,doi.org/10.3390/cancers15235626, available via: https://www.mdpi.com/2072-6694/15/23/5626 CC BY 4.0.
Well-Differentiated Systemic Mastocytosis (WDSM):
WDSM is a rare variant with mature-appearing mast cells that often lack the typical KIT D816V mutation. Its clinical behavior and biology remain poorly defined.
Etiology & Pathophysiology of Systemic Mastocytosis
The primary cause of systemic mastocytosis is an acquired mutation in the KIT gene, most commonly KIT D816V, leading to continuous activation of the KIT receptor tyrosine kinase and overproduction of abnormal mast cells. Other gene mutations, including SRSF2, ASXL1, and RUNX1, may occur in advanced cases or those with associated hematologic neoplasms.
Mast cells are immune cells present in connective tissues and are activated by both IgE-mediated and non-IgE mechanisms, functioning as effector cells in allergic and hypersensitivity reactions. The KIT gene mutations are not inherited. The constant signalling from the mutated KIT protein results in the excessive accumulation of mast cells in various organs, disrupting their normal function.[3]
Risk Factors
Some of the factors that trigger the mast cell activation for developing systemic mastocytosis are:
- Spices
- Alcohol
- Insect bites
- Certain medications (NSAIDs, anesthesia, muscle relaxers)
- Stress
- Sudden temperature changes in your body
Signs & Symptoms of Systemic Mastocytosis
Systemic mastocytosis involves a variety of symptoms (it affects various organ systems). The common symptoms include:
Allergic and Cutaneous Symptoms
- Anaphylaxis (severe allergic reactions) that are triggered by numerous stimuli, including insect stings, stress, certain foods, alcohol, certain medicines, physical exertion, and temperature changes.
- Itching (pruritus)
- Flushing
- Stinging
- Reddish-brown raised patches or urticaria pigmentosa
Gastrointestinal Symptoms
These include:
- Vomiting
- Abdominal pain
- Diarrhea
- Nausea
- Bloating
- Gastroesophageal reflux
Musculoskeletal symptoms
- Muscle pain
- Bone pain
- Osteoporosis
- Pathological fractures
- Osteopenia
Cardiovascular symptoms
- Hypotension
- Vasodilation
- Rapid heart rate
- Light headedness
- Dizziness
Neurological and psychiatric symptoms
- Brain fog
- Anxiety
- Syncope
- Depression
- Mood swings
- Headaches
Respiratory symptoms
- Shortness of breath
- Nasal congestion
- Throat swelling
- Wheezing
Systemic symptoms
- Fatigue
- Enlarged liver, spleen, and lymph nodes
- Bleeding disorders
- Anemia
- Organ dysfunction in most severe cases.
Diagnosis of Systemic Mastocytosis
The healthcare provider begins with a physical exam and proceeds from there.
Physical Examination & History:
Physical exam findings in patients with systemic mastocytosis include signs of anemia, such as pallor. Patients may also present with lymphadenopathy, splenomegaly, and hepatomegaly. Some may present gastrointestinal bleeding, thrombocytopenia, hypersplenism, signs of urticaria, osteolysis, and, rarely, pathological fractures. Healthcare providers note the previous history of anaphylaxis to any specific food, medication, or other triggers. Additional coexistent hematologic disorders, such as non-Hodgkin lymphoma, Castleman disease, and monoclonal gammopathy, can also be present.
Diagnostic Workup
- Complete blood count and liver/renal function tests
- Serum tryptase level (often >20 ng/mL)
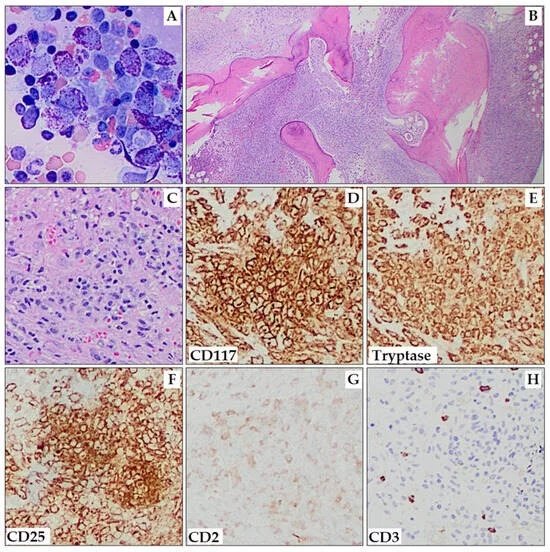
- Bone marrow aspiration and biopsy with immunophenotyping (CD25, CD2)
- Detection of KIT D816V mutation via PCR or next-generation sequencing
- Bone densitometry for assessing osteoporosis
Additional Diagnostic Options:
Based on the clinical presentation, these additional tests may be required:
- Abdominal computed tomography scan for abnormal white blood cell counts, lymphadenopathy, and hepatosplenomegaly.
- For gastrointestinal symptoms, clinicians can perform biopsies and gastrointestinal endoscopy.
- Liver function tests.[4]
Criteria for Diagnosis of Systemic Mastocytosis:
According to the World Health Organization (WHO), systemic mastocytosis is diagnosed when 1 major and 1 minor criterion, or at least 3 minor criteria, are met.
Minor Criteria
- Presence of atypical mast cells (greater than 25%)
- Presence of KITD 816V mutation
- Abnormal mast cell CD25 expression
- Elevated serum tryptase level (above 20 ng/ml)[5]
Major Criteria
Presence of multifactorial clusters of abnormal mast cells (more than 15% of mast cells in clusters) in the extracutaneous tissues or bone marrow.[6]
Differential Diagnosis
Differential diagnosis is broad for systemic mastocytosis. These include:
- Acute urticaria (itchy, raised skin welts, without mast cell infiltration)
- VIPoma (rare endocrine tumor from the pancreas, more severe than SM)
- Carcinoid syndrome (paraneoplastic syndrome, unlike SM, it lacks mast cell aggregates or KIT mutations)
- Zollinger-Ellison syndrome (growth of tumors in the pancreas or small intestine, but does not involve mast cells)
- Myeloproliferative disorder (a group of blood disorders, but no KIT-activating mutations)
- Malabsorption (without mast cell infiltration)
- Inflammatory or irritable bowel diseases (no mast cell accumulation).
Management & Treatment of Systemic Mastocytosis
Management and treatment of systemic mastocytosis can be tailored to be symptom-specific and type-specific.
Symptom-Specific Management:
Healthcare providers consider the following management options for symptom-specific systemic mastocytosis.
- Initial pharmacological management includes the use of H1 and H2 antihistamines. Antihistamine H1 to prevent flushing and itching, while H2 antihistamine for heartburn, diarrhea, cramping, and abdominal pain.
- Patients who experience recurrent anaphylaxis with hemodynamic instability should also get injections of epinephrine along with maximum doses of H1 and H2 antihistamines and anti-leukotriene drugs.[7]
- Omalizumab for managing persistent anaphylaxis.[8]
- Doctors recommend antileukotriene agents as optional treatment for abdominal cramping, itching, and flushing for patients unresponsive to antihistamines.
- Patients with persistent flushing require aspirin (in non-asthmatic patients).
- Doctors consider oral cromolyn sodium, proton pump inhibitors, and glucocorticoids for systemic mastocytosis with severe malabsorption or ascites.[9]
- Daily intakes of Vitamin D and calcium for patients with osteoporosis-related fractures. Bisphosphonates (pamidronate or zoledronate) are also helpful.
- For patients who are unresponsive to anti-mediator therapies omalizumab, doctors consider low-dose interferon-alpha or glucocorticoids for refractory systemic symptoms
Type–Specific Management:
ISM or SSM
Cytoreductive therapy for those who suffer from recurrent anaphylaxis (uncontrolled with anti-mediator treatments).[10]
Aggressive SM
Management options include midostaurin, cladribine, TKI inhibitors, or interferon-alpha, depending on the patient’s mutation status, along with a bone marrow transplant. Treatment of this advanced stage involves mitigating organ dysfunction and improving the quality of life.
SM-AHN
Treatment includes managing the associated hematopoietic disorder. Doctors consider hematopoietic cell transplantation (HCT) after initial salvage therapy in extreme cases.
Mast-Cell Leukemia
Managed with cytoreductive therapy and, in selected cases, hematopoietic stem cell transplantation, though prognosis remains poor.
Prognosis
There is no definitive cure for systemic mastocytosis. Indolent SM has a near-normal life expectancy, whereas ASM, SM-AHN, and MCL carry a poor prognosis. Bone marrow transplantation may offer long-term remission in aggressive disease, but it is not a standard therapy.[11]
Complications
If left untreated, this condition can lead to several disorders. These include:
- Anemia and coagulopathy
- Hypereosinophilic syndrome
- Non-Hodgkin lymphoma or monoclonal gammopathy
- Hepatosplenomegaly and organ failure
- Recurrent anaphylaxis
Final Remarks
The unique clinical profile of systemic mastocytosis, combined with advancements in diagnostic and pharmacological approaches, poses significant health challenges. Close coordination of care among allergists, immunologists, and hematologists is crucial for preventing the progression of the disease and avoiding misdiagnosis. In the event of anaphylactoid reactions, patients should always carry epinephrine-filled syringes. Patients should learn to administer epinephrine to themselves in emergency situations.
References
[1] Kevin, Y. T., Chen, W., Puttock, E. J., Chowdhury, S., Miller, K., Powell, D., … & Zeiger, R. S. (2024). MASTering systemic mastocytosis: Lessons learned from a large patient cohort.Journal of Allergy and Clinical Immunology: Global,3(4), 100316.
[2] Gangireddy, M., & Ciofoaia, G. A. (2019). Systemic mastocytosis.
[3] Abid, A., Malone, M. A., & Curci, K. (2016). Mastocytosis.Primary Care: Clinics in Office Practice,43(3), 505-518.
[4] Scherber, R. M., & Borate, U. (2018). How we diagnose and treat systemic mastocytosis in adults.British journal of haematology,180(1), 11-23.
[5] Desmond, D. H., & Carmichael, M. G. (2018). Systemic mastocytosis: the difficult patient with a rare disease. Case presentation and brief review.Hawai’i Journal of Medicine & Public Health,77(2), 27.
[6] Desmond, D. H., & Carmichael, M. G. (2018). Systemic mastocytosis: the difficult patient with a rare disease. Case presentation and brief review.Hawai’i Journal of Medicine & Public Health,77(2), 27.
[7] Tolar, J., Tope, W. D., & Neglia, J. P. (2004). Leukotriene-receptor inhibition for the treatment of systemic mastocytosis.New England Journal of Medicine,350(7), 735-736.
[8] Jagdis, A., & Vadas, P. (2014). Omalizumab effectively prevents recurrent refractory anaphylaxis in a patient with monoclonal mast cell activation syndrome.Annals of Allergy, Asthma & Immunology,113(1), 115-116.
[9] Valent, P., Sperr, W. R., & Akin, C. (2010). How I treat patients with advanced systemic mastocytosis.Blood, The Journal of the American Society of Hematology,116(26), 5812-5817.
[10] Laroche, M., Livideanu, C., Paul, C., & Cantagrel, A. (2011). Interferon alpha and pamidronate in osteoporosis with fracture secondary to mastocytosis.The American journal of medicine,124(8), 776-778.
[11] Abid, A., Malone, M. A., & Curci, K. (2016). Mastocytosis.Primary Care: Clinics in Office Practice,43(3), 505-518.