
 syndrome, is a rare, genetic (inherited) disorder that...){kind=link}

Von Hippel Lindau Von Hippel-Lindau disease, also known as Von Hippel-Lindau (VHL) syndrome, is a rare, genetic (inherited) disorder that causes cystic and tumorous growths throughout the body. The multi-system disease is not characterized by universal spread; most lesions, such as hemangioblastomas and cysts, are benign, while clear-cell renal cell carcinoma (ccRCC) and some pancreatic neuroendocrine tumors carry the main risk of metastasis. The estimated prevalence of this genetic disorder is 1/30,000 to 1/50,000 individuals. The average incidence of the genetic disorder is approximately 1 in 36,000 live births, with males and females equally affected.
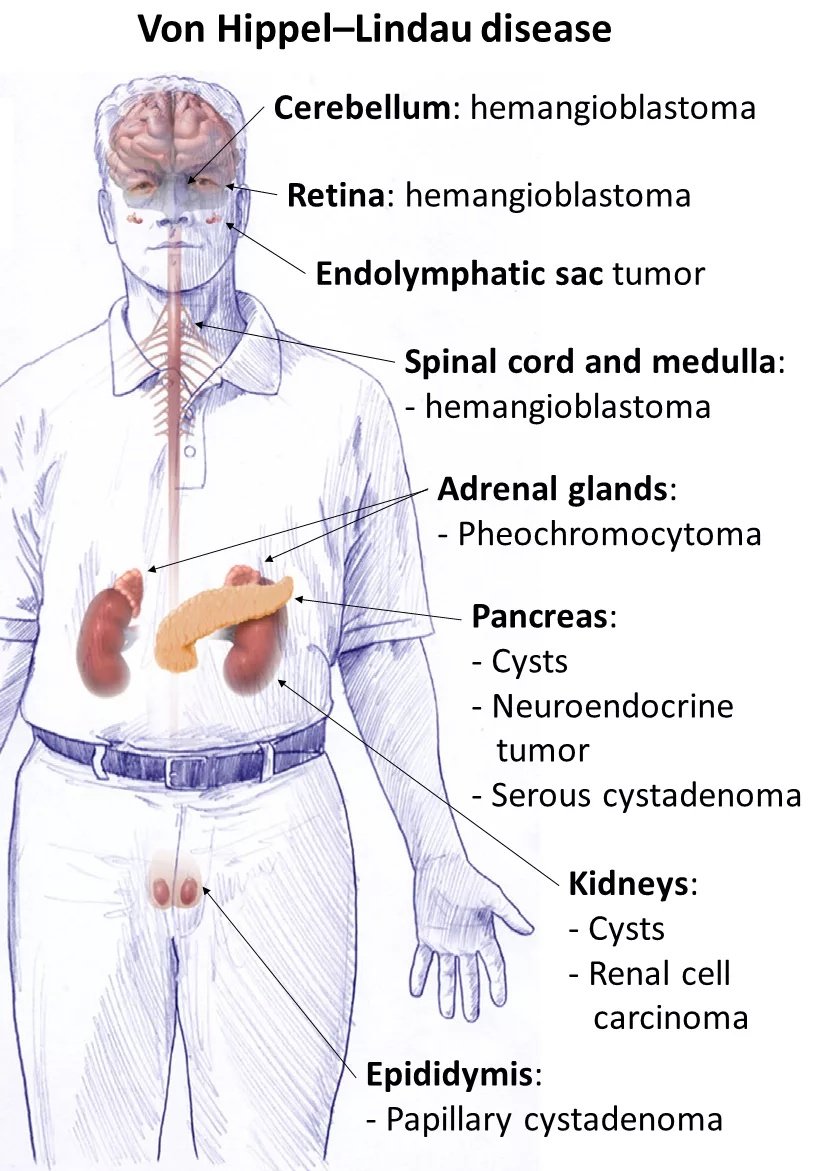
Von Hippel-Lindau disease causes the development of characteristic hemangioblastomas, which are vascular tumors that grow in the brain, spinal cord, and retina. These tumors are benign but can cause significant symptoms by pressing on nearby structures. The mainstay of the treatment is surgical removal of the tumors. However, newer targeted therapies such as belzutifan are now transforming treatment options.
Types Of Cysts: Von Hippel Lindau
Cysts are fluid-filled cavities that develop due to some underlying pathology. In this case, a genetic mutation increases the chances of cyst formation in patients. The common locations for VHL-induced cysts are:
Kidneys:
Renal manifestations are not uncommon in VHL patients. While cancers like renal cell carcinoma are frequent, patients may also suffer from multiple renal cysts at the same time. Patients may develop numerous bilateral cysts, which can compromise kidney function and occasionally extend into adjacent structures. Bilateral kidney cysts can negatively affect the quality of life by impacting both kidneys.
Pancreas:
Cysts can also make your pancreas their permanent abode. Oftentimes, patients visit clinics with simultaneous problems in the kidneys and the pancreas. In one clinical study, a 49-year-old woman presented to the ER with jaundice and pruritus. Imaging scans of the patient revealed bilateral renal cysts accompanied by numerous pancreatic cysts. The multitude of abnormal fluid-filled structures in the pancreas gave a “honeycomb appearance,” and she was diagnosed with Von Hippel-Lindau disease.
Types Of Tumors/Cancers
As already mentioned, VHL significantly increases the patient’s propensity to develop different types of cancers. The most frequently associated tumors and cancers include:
Hemangioblastomas:
The most common non-cancerous, tumorous growth linked to VHL is hemangioblastomas. They are benign vascular tumors of the central nervous system (brain, spinal cord) and retina. CNS hemangiomas can have varied neurological presentations and may present with accelerated tumor growth. Hemangioblastomas of the cerebellum and brainstem are present in a large number of patients.

An ocular angiogram of a Von Hippel-Lindau disease patient reveals a retinal hemangioblastoma.
Retinal hemangioblastomas are common manifestations of VHL disease. The disease can cause multiple hemangioblastomas in the blood vessels of the eye, which eventually lead to ophthalmic complications like vitreous hemorrhage, retinal detachment, glaucoma, and even permanent vision loss.
Healthcare providers have identified various types of ophthalmic hemangioblastomas associated with von Hippel-Lindau disease, including hemangioblastomas of the optic nerve and chiasm.
Clear Cell Renal Cell Carcinomas (ccRCC):
It is the most common type of kidney cancer that is frequently associated with Von Hippel-Lindau disease. ccRCC is the leading cause of cancer-related death in VHL patients. Reports suggest that approximately two-thirds of VHL patients present with multiple clear cell renal carcinomas and cysts throughout their lives. The occurrence of kidney cancers is attributed to germline mutations. Clinical research shows that RCC has a high rate of disease progression and metastasis development. Most VHL patients succumb to life due to metastasis from renal clear cell carcinomas. Nephron-sparing surgery (partial nephrectomy) is the preferred treatment, and many centers intervene once tumors reach ~3 cm to balance cancer control with preservation of kidney function.
Pancreatic Neuroendocrine (PanNETs) Cancers:
Pancereatic cysts are common findings of the syndrome. Cysts and cancerous growths of the insulin-producing organ can co-exist, making management difficult. These tumors are rarely found in nature and develop in the endocrine cells of your pancreas. However, they develop frequently in VHL patients. Researchers believe that tumor size is the strongest predictor of metastatic potential, with those >3 cm carrying a higher risk.. Large tumors tend to spread to other organs.

The image shows a pancreatic neuroendocrine tumor on H&E and Pap stains in a Von Hippel-Lindau disease patient.
The occurrence of PanNETs is associated with a high mortality rate, which is why clinicians advise active surveillance of high-risk patients.
Pheochromocytomas:
Another type of rare cancer associated with Von Hippel-Lindau syndrome is pheochromocytoma. This treatable tumor grows in the adrenal gland (adrenal medulla) and is a hallmark feature of VHL disease. Clinicians classify VHL disease based on pheochromocytomas.
These tumors are frequently found in VHL patients along with paragangliomas. A systematic review suggests that pheochromocytomas and/or paragangliomas (PPGLs) are common manifestations of the Von Hippel-Lindau disease. The benign tumor causes undulations in the blood sugar and blood pressure levels and alters the body’s response to stress.
Endolymphatic Sac Tumors:
Patients suffering from Von Hippel-Lindau disease can also develop locally invasive tumors of the endolymphatic duct (within the temporal bone). It is an uncommon epithelial tumor that may develop in your inner ear. An early diagnosis and treatment of this very neoplasm is crucial, as it can lead to permanent hearing loss in both ears. The incidence of this slow-growing tumor is low and found to lie between 4% and 16%. Bilateral endolymphatic sac tumors are indicative of VHL. Symptoms of the benign growth include vertigo, tinnitus, and hearing loss etc.
It was found in a systematic review that profound hearing loss and facial impairment (due to facial nerve palsy) were the most serious clinical complications entailed by VHL-induced endolymphatic sac tumors.
Cystadenomas:
A small number of patients also suffer from broad ligament cystadenomas. These tumors are present in males and females. In males, papillary cystadenomas are found in the epididymis (tube connected to the testes), while in female patients, cystadenomas are present in the ovaries and fallopian tubes. As the disease is more common in men, broad ligament cystadenomas (of the ovaries) are rarer than cystadenomas of the epididymis.
Von Hippel-Lindau Classification
Based on the presence of different types of cancers, healthcare professionals divide VHL into the following categories:
Type 1:
This is allocated to all VHL cases that do not have pheochromocytomas.
Type 2:
Pheochromocytoma is present in patients and is further classified into:
- 2A: CNS hemangioblastomas are present with pheochromocytoma but not RCC.
- 2B: Both CNS hemangioblastomas and RCCs are present with pheochromocytoma.
- 2C: No hemangioblastomas or RCCs are found with pheochromocytoma.
Von Hippel-Lindau Disease Symptoms
Symptoms of the disorder vary according to the location and size of the tumor. In general, symptoms like headache, nausea, and vomiting are common to the majority of cancers/tumors. Based on the organs involved, the most prevalent symptoms of Von Hippel-Lindau disease include:
The illustration shows common symptoms of Von Hippel-Lindau disease.
Neurological Symptoms:
Tumors in different parts of the CNS can press on the neighboring structures, leading to neurological symptoms. Patients with brain and spinal cord hemangioblastomas complain of nausea, vomiting, and headaches, which may be attributed to raised intracranial pressure. Impinging on nerves may also present with muscle weakness, dizziness, gait issues (loss of balance), and bowel/bladder dysfunctions, etc.
Ophthalmic Symptoms:
As retinal hemangioblastomas are fairly common, patients encounter different eye symptoms. The retinal angiomas present with floaters and vision impairment. If left untreated, the disease progresses to significant visual morbidity and problems like retinal detachment. The result of this is blindness. Thus, screening, timely diagnosis, and treatment are crucial.
Kidney Symptoms:
In the vast majority of cases, renal clear cell carcinomas are asymptomatic. However, when present, patients experience issues like blood in urine (hematuria) and pain between the ribs and hips (flank pain). Studies show that some patients complain of palpable abdominal mass, flank pain, and hematuria due to VHL-induced ccRCC.
Hearing Issues:
Endolymphatic sac tumors (ELSTs) of the inner ear can present with hearing difficulties and balance issues in patients. Several patients complain of tinnitus (bell/ringing in the ear) and loss of hearing. The tumors can also impact a patient’s ability to maintain balance. Thus, vertigo is also a feature of ELSTs.
Blood Pressure Irregularities:
Pheochromocytomas are known to cause disturbances in the body’s fight or flight response. The tumors in the adrenal medulla have direct implications for the patient’s blood pressure. Many patients suffer from raised blood pressure, i.e., hypertension. Based on clinical studies, common symptoms of this particular type of tumor include headache, palpitations, paroxysmal hypertension, and sweating.
Von Hippel-Lindau Disease Causes
The Von Hippel-Lindau gene (VHL gene) is a tumor suppressor gene that ensures normal division of cells. It is present on chromosome 3p25-26 and prevents cells from dividing uncontrollably. Mutations in the VHL gene lead to uncontrolled division and the development of different types of tumors/cancers throughout the body. With the help of radiogenomics, clinicians have identified VHL gene mutations in different types of VHL-induced cancers, like clear cell renal cell carcinomas. About 80–85% of cases are inherited from an affected parent, while 15–20% occur de novo.
Von Hippel-Lindau Disease Inheritance:
The disorder is transferred from parents to offspring in an autosomal dominant pattern. This means only one copy of the mutated gene can induce the disease. You can be affected with the syndrome if one of your biological parent passes the abnormal VHL gene to you. Thus, the chances of a child getting Von Hippel-Lindau syndrome if one of the parents passes the mutated gene are 50%.
Von Hippel-Lindau Disease Diagnosis
History Taking & Examination:
Diagnosing genetic conditions is a little tricky. Your healthcare provider will start by taking a history of your symptoms. In the vast majority of cases, frequent and multiple hemangioblastomas and clear cell carcinomas in a patient raise concerns. If VHL disease is suspected, your doctor will ask about your family history of Von Hippel-Lindau disease. This may be followed by a physical examination to notice unusual changes in the body induced by cysts, tumors, and cancers.
Genetic Testing:
A conclusive diagnosis is made via genetic testing. This involves identifying the mutated genes that are culprits. This gold standard of testing for VHL disease involves taking a sample from the patient (buccal swab or blood sample, etc.). Pathologists then search for specific (suspected) DNA mutations in molecular testing. This diagnostic test is indicated in VHL and VHL-associated tumors and yields good diagnostic accuracy.
Imaging Studies:
As the disease is notorious for causing an array of cancers in different parts of the body, imaging studies play a crucial part in determining the location and severity of the tumor. Freuqently used imaging tests in modern clinics include MRI scans and CT scans.
Clinical Evaluations:
Mostly, large tumors/cancers interfere with the normal abilities of the patients. Your doctor may carry out the following evaluations to check the extent of damage caused by the cancerous growths:
- Eye test: Ophthalmic examination is important because of the high prevalence of retinal hemangioblastomas. Tests include optical coherence tomography (OCT) and ultrasonography.
- Hearing tests: Doctors check for any hearing loss due to endolymphatic sac tumors. Audiometry tests, like pure-tone audiometry, may be adopted.
Von Hippel-Lindau Disease Treatment
There is no way to correct the genetic abruptions. However, doctors provide symptomatic management to patients with tumors and cysts. The treatment modality depends on the type, location, and size of the growth (tumor/cyst). Doctors usually adopt the following strategies:
Surgery:
Surgical excision of the tumor/cyst is the primary method of managing VHL-induced tumors. Conventionally, doctors have utilized surgical intervention as the ultimate way of treating VHL tumors. Clinical studies suggest that surgical removal of retrobulbar hemangioblastomas is a safe and effective procedure.
Gamma-knife surgery is used to remove small, solid tumors (hemangioblastomas). This technique also helps in excising tumors present in inoperable sites. Early nephrectomy (surgical removal of the kidneys) is the ideal treatment option for renal cell carcinomas.
Surgeons remove pheochromocytomas surgically. In pediatric patients, partial adrenalectomy (a part of the adrenal gland is removed) is the treatment of choice. While surgical excision seems like the ideal way of management, doctors avoid excising pancreatic neuroendocrine tumors. Only those tumors PENnets are removed that are greater than 3 cm, or have a high risk of metastasis, or have a doubling rate of fewer than 500 days. Early removal of endolymphatic sac tumors helps avoid hearing loss in VHL disease patients.
However, modern studies reveal that surgery negatively affects patients’ quality of life and worsens their mental health. Therefore, other modalities need to be adopted.
Targeted Therapy:
Thanks to medical advancements, now, doctors can administer specialized drugs that target only the tumor cells while keeping the healthy body cells safe. Peptide receptor radionuclide therapy and Tyrosine kinase inhibitor drugs have shown promising results in the management of different types of cancers.
Belzutifan is an HIF-2ɑ inhibitor (hypoxia-inducible factor 2 alpha protein) that works by blocking the activity of the HIF-2ɑ protein. This very protein is overexpressed in cancer cells. By targeting the protein activity, the drug has shown good results in the management of VHL-associated cancers and has low toxicity, with only reported side effects being fatigue and anemia.
Radiotherapy:
Oncologists find radiation therapy to be a useful modality for different cancers associated with Von Hippel-Lindau disease. This type of cancer management works best for hemangioblastomas (CNS and retinal) and renal cell carcinomas. Most of the time, doctors shift to radiotherapy when surgery is not a feasible option or when other strategies have failed. Types of radiation therapies used for VHL tumors include external beam radiation and stereotactic radiosurgery. A prospective trial revealed that stereotactic body radiotherapy (SBRT) has an excellent tumor control rate. It was well tolerated by patients and helped preserve kidney function in patients with multiple renal cysts.
Hormone Therapy:
This type of therapy can be used in tumors that alter the hormonal levels of the body. Sometimes, doctors use hormone therapy to address pancreatic neuroendocrine tumors and pheochromocytomas.
Chemotherapy:
Chemical systemic therapy is not the preferred treatment modality, but it may be employed in the management of renal cell carcinomas and pancreatic cancers.
Doctors may also adopt the following minimally invasive procedures to manage extensive hemangioblastomas:
- Diathermy
- Cryocoagulation
- Cryoablation and radiofrequency ablation
- External beam radiotherapy
- Laser therapy
Von Hippel-Lindau Disease Prognosis
The prognosis for Von Hippel-Lindau disease varies, depending on the type of cancer. Improved screening and advanced treatment options (especially targeted therapy) have significantly improved prognoses.
Von Hippel-Lindau Disease Life Expectancy
According to a study, the estimated mean life expectancies for male and female patients of VHL disease were 67 and 70 years, respectively. Doctors conclude that surveillance of individuals with the VHL gene mutation proves to be beneficial. Over time, the frequency of renal cell carcinoma and VHL-associated deaths has decreased.
Von Hippel-Lindau Screening
Doctors advise diagnostic audiogram-based screening of patients between the ages of 11 years and 65 years to prevent aggravation of symptoms, as this age bracket carries a high risk of inner ear tumors.
Final Word
Von Hippel-Lindau is a rare, inherited disorder that causes the development of cysts (in the pancreas and kidneys), benign tumors of the lungs, hemangioblastomas of the CNS and eye, and cancers such as renal cell carcinoma, pancreatic neuroendocrine tumors, pheochromocytomas, and endolymphatic sac tumors. Symptoms vary according to the type and location of the tumor. However, common presentations include nausea, vomiting, headache, vision impairment, hearing loss, and blood pressure abnormalities.
The occurrence of the disease is attributed to a mutation in the Von Hippel-Lindau gene (a tumor suppressor gene). Doctors diagnose the condition with physical examination, family history, and genetic testing. Conventionally, the mainstay of treatment was surgical removal of cysts and cancers. However, now advanced targeted therapies with drugs like Belzutifan have shown much better results with minimal side effects.
References
[1] Mikhail, M. I., & Singh, A. K. (2017). Von Hippel-Lindau Syndrome.
[2] Maher, E. R., Neumann, H. P., & Richard, S. (2011). von Hippel–Lindau disease: A clinical and scientific review.European Journal of Human Genetics,19(6), 617-623.
[3] Kirste, S., Rühle, A., Zschiedrich, S., Schultze-Seemann, W., Jilg, C. A., Neumann-Haefelin, E., … & Kim, E. (2022). Stereotactic Body Radiotherapy for Renal Cell Carcinoma in Patients with Von Hippel–Lindau Disease—Results of a Prospective Trial.Cancers,14(20), 5069.
[4] Wang, X., Xu, Z., & Huang, W. F. (2023). An Amazing Image of Von Hippel–Lindau Syndrome.Indian Journal of Surgery,85(2), 442-443.
[5] Takami, H., Graffeo, C. S., Perry, A., Brown, D. A., Meyer, F. B., Burns, T. C., & Parney, I. F. (2022). Presentation, imaging, patterns of care, growth, and outcome in sporadic and von Hippel–Lindau-associated central nervous system hemangioblastomas.Journal of neuro-oncology,159(2), 221-231.
[6] Huang, Y., Hu, W., & Huang, X. (2022). Retinal hemangioblastoma in a patient with Von Hippel-Lindau disease: a case report and literature review.Frontiers in Oncology,12, 963469.
[7] Vergauwen, E., Klingler, J. H., Krüger, M. T., Steiert, C., Kuijpers, R., Rosahl, S., … & Gläsker, S. (2024). Optic nerve and chiasm hemangioblastomas in von Hippel-Lindau disease: report of 12 cases and review of the literature.Frontiers in Oncology,14, 1334564.
[8] Prabhaswari, L., & Adnyana, I. W. L. (2024). Patient with Von Hippel-Lindau disease diagnosed with Renal Cell Carcinoma.Research, Society and Development,13(12), e07131247548-e07131247548.
[9] Penitenti, F., Landoni, L., Scardoni, M., Piredda, M. L., Cingarlini, S., Scarpa, A., … & Davi, M. V. (2021). Clinical presentation, genotype–phenotype correlations, and outcome of pancreatic neuroendocrine tumors in Von Hippel–Lindau syndrome.Endocrine,74(1), 180-187.
[10] Arnon, L., Halperin, R., & Tirosh, A. (2021). Impact of pancreatic neuroendocrine tumor on mortality in patients with von Hippel-Lindau disease.Endocrine Practice,27(10), 1040-1045.
[11] Castro-Teles, J., Sousa-Pinto, B., Rebelo, S., & Pignatelli, D. (2021). Pheochromocytomas and paragangliomas in von Hippel–Lindau disease: not a needle in a haystack. Endocrine connections, 10(11), R293-R304.
[12] Christopher, L. H., Lekovic, G. P., & Mehta, G. U. (2024). Endolymphatic Sac Tumors in von Hippel-Lindau Disease. InVon Hippel-Lindau Disease: A Comprehensive Guide to Diagnosis, Treatment, and Management(pp. 125-140). Cham: Springer International Publishing.
[13] Gioacchini, F. M., Kaleci, S., Chiarella, G., Viola, P., Pisani, D., Scarpa, A., … & Re, M. (2022). Symptoms and clinical features in patients affected by endolymphatic sac tumor: a systematic review and meta-analysis.European Archives of Oto-Rhino-Laryngology,279(11), 5081-5088.
[14] Bergaoui, H., Ghaddab, I., Dhouib, W., Njima, M., Belghaib, I., Farhat, I. B., … & Faleh, R. (2025). Bilateral papillary cystadenoma of the broad ligament: a manifestation of Von Hippel–Lindau disease: a case report.Journal of Medical Case Reports,19(1), 159.
[15] Jankovic, D., Selimovic, E., Kuharic, M., Splavski, B., Rotim, K., & Arnautovic, K. I. (2024). Understanding adult central nervous system hemangioblastomas: a systematic review.World Neurosurgery,191, 119-127.
[16] Milam, R., & Daniels, A. B. (2024). Retinal hemangioblastomas. InVon Hippel-Lindau Disease: A Comprehensive Guide to Diagnosis, Treatment, and Management(pp. 61-104). Cham: Springer International Publishing.
[17] Bahadoram, S., Davoodi, M., Hassanzadeh, S., Bahadoram, M., Barahman, M., & Mafakher, L. (2022). Renal cell carcinoma: an overview of the epidemiology, diagnosis, and treatment.G Ital Nefrol,39(3), 2022.
[18] Anyfanti, P., Mastrogiannis, Κ., Lazaridis, Α., Tasios, Κ., Vasilakou, D., Kyriazidou, Α., … & Gkaliagkousi, Ε. (2023). Clinical presentation and diagnostic evaluation of pheochromocytoma: case series and literature review.Clinical and Experimental Hypertension,45(1), 2132012.
[19] Greco, F., D’Andrea, V., Beomonte Zobel, B., & Mallio, C. A. (2024). Radiogenomics and texture analysis to detect von Hippel–Lindau (VHL) mutation in clear cell renal cell carcinoma.Current Issues in Molecular Biology,46(4), 3236-3250.
[20] Dwivedi, A., Moirangthem, A., Pandey, H., Sharma, P., Srivastava, P., Yadav, P., … & Mandal, K. (2022). Von Hippel–Lindau (VHL) disease and VHL-associated tumors in Indian subjects: VHL gene testing in a resource constraint setting.Egyptian Journal of Medical Human Genetics,23(1), 126.
[21] Alvarez, R., Mastorakos, P., Hogan, E., Scott, G., Lonser, R. R., Wiley, H. E., … & Chittiboina, P. (2021). Retrobulbar hemangioblastomas in von hippel-lindau disease: clinical course and management.Neurosurgery,88(5), 1012-1020.
[22] Sundaram, M., Atkinson, C., Cooper, C., Taylor-Stokes, G., Mann, J., & Iliopoulos, O. (2025). The impact of surgery on patients with von Hippel-Lindau-associated tumors: an international patient survey.The Oncologist, oyaf206.
[23] Curry, L., & Soleimani, M. (2024). Belzutifan: a novel therapeutic for the management of von Hippel–Lindau disease and beyond.Future Oncology,20(18), 1251-1266.
[24] Kirste, S., Rühle, A., Zschiedrich, S., Schultze-Seemann, W., Jilg, C. A., Neumann-Haefelin, E., … & Kim, E. (2022). Stereotactic Body Radiotherapy for Renal Cell Carcinoma in Patients with Von Hippel–Lindau Disease—Results of a Prospective Trial.Cancers,14(20), 5069.
[25] Binderup, M. L. M., Jensen, A. M., Budtz-Jørgensen, E., & Bisgaard, M. L. (2017). Survival and causes of death in patients with von Hippel-Lindau disease.Journal of medical genetics,54(1), 11-18.
[26] Mehta, G. U., Kim, H. J., Gidley, P. W., Daniels, A. B., Miller, M. E., Lekovic, G. P., … & Lonser, R. R. (2022). Endolymphatic sac tumor screening and diagnosis in von Hippel-Lindau disease: a consensus statement.Journal of Neurological Surgery Part B: Skull Base,83(S 02), e225-e231.