
{kind=link}

Wilson’s disease Wilson’s disease (WD) is a rare inherited disease that involves a mutation (changes) in the gene ATP7B, which encodes a copper-transporting ATPase involved in copper metabolism. This disease is characterized by the toxic accumulation of copper in the body, especially in the liver, eye, and brain, presenting with a wide range of hepatic, neurological, and psychiatric symptoms. Early diagnosis and management are crucial, followed by lifelong therapy with chelating agents to prevent the accumulation of copper in the system.
Epidemiology of WD: Wilson’s disease
The estimated prevalence in many populations is around 1 in 30,000. The prevalence can vary by population and genetic factors. Approximately 1% of people carry a heterozygous (single) gene mutation that they can unknowingly pass on to their offspring.
It affects men and women equally, since it is an inherited disease. Family history of diagnosed WD, especially a sibling, is a key risk factor that you may also have WD.
Causes and Inheritance of Wilson’s disease
WD is caused by mutations or changes in the ATP7B gene. This mutated gene is inherited from the parents to offspring, putting them at risk of developing WD. Being an autosomal-recessive disorder, the mutated gene needs to be passed by both parents for the child to show symptoms of the disease. If both parents are carriers, there is a 25% chance the child will develop WD, a 50% chance they will be a carrier, and a 25% chance they will be unaffected. If both parents have Wilson’s disease, all children will inherit the condition.
Pathophysiology of Wilson’s disease
Wilson’s disease is a genetic disorder affecting the ATP7B gene, which is responsible for encoding a copper-transporting ATPase enzyme in hepatocytes. This protein is responsible for delivering and incorporating copper into apoceruloplasmin and forming ceruloplasmin. When the incorporation of dietary copper into ceruloplasmin is ineffective, it leads to excess buildup. In normal conditions, the copper is bound to ceruloplasmin, which would then manage the distribution of copper into different parts of the body for essential functioning. With reduced or impaired ceruloplasmin formation, copper starts accumulating in the body, primarily in the liver and the brain. Defective ATP7B impairs copper incorporation into ceruloplasmin and biliary excretion. Accumulation of copper in the liver leads to liver damage, and excess levels in the brain result in neurological and psychological symptoms.
Symptoms of Wilson’s disease
Symptoms can be categorized by onset and affected organs.
Early symptoms
These symptoms are general, including tiredness, loss of appetite, tremors, and changes in personality or mood.
Eye manifestations
Copper accumulation in the eye forms“Kayser-Fleischer” rings. They are brownish-yellow or copper colored rings that form on the outer edge of the colored part of the eye (iris). The Kayser-Fleischer rings usually appear first at the top and bottom of the iris, and eventually form a full circle. Initially, the ophthalmologist can only see the rings under a special light called a “slit-lamp.” It may be visible to the naked eye in cases of severe copper accumulation. They are asymptomatic and do not cause any trouble, and may diminish with decreasing copper levels. Moreover, they are helpful as an indicator of disease progression and the effectiveness of treatment.
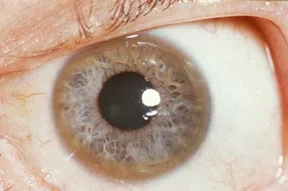
Kayser–Fleischer ring is seen as a golden-brown ring around the edge of the cornea, caused by copper buildup in the eye. Image via:Wikimedia Commons. Licensed underCC BY-SA 3.0.
Neurological manifestations6Jopowicz A, Tarnacka B. Neurological Wilson’s Disease Signs—Hepatic Encephalopathy or Copper Toxicosis? Diagnostics. 2023 Feb 27;13(5):893.
Copper builds up in the parts of the brain controlling movement, i.e., the basal ganglia, brainstem, thalamus, etc. Damage to these areas leads to related signs and symptoms:
- Tremors: “Wing-beating” tremor is characteristic of Wilson’s disease. When arms are held forward and stretched sideways, the tremor looks like a bird’s wing beating. It is involuntary, persistent, and significantly visible.
- Dystonia: Dysfunction of the muscles can lead to involuntary and repeated muscle contractions or loss of tone in them. This dysfunction could either affect the whole body or only specific muscles, e.g., the facial muscles.
- Dysarthria: Difficulty in speaking is very common. Speech is imprecise, slow, weak, and uncoordinated.
- Dysphagia: Difficulty in swallowing
- Ataxia: Patient is unsteady while walking, with clumsy movements. There are abnormalities in posture and body coordination.
- Seizures: A study showed that 14.5% of people with WD developed seizures.
- Mood or personality changes can include irritability, depression, and insomnia, and can even lead to psychosis.
Liver symptoms
In Wilson’s disease, copper accumulation most commonly affects the liver. As copper accumulates in the liver, it causes severe liver damage and eventually leads to liver failure. This shows up as:
- Jaundice: Yellowing of the skin and eyes.
- Fatigue, nausea, abdominal pain.
- Ascites: Accumulation of fluid in the abdominal cavity.
- Easy bruising
- Frequent bleeding (such as nosebleeds)
- Dark urine
Other
Buildup of copper in your body also shows up as:
- Blue fingernails or toenails (also known as azure lunulae)
- Skeletal abnormalities (such as arthritis, osteoporosis)
- Cardiac involvement (such as rhythm abnormalities)
- Loss of sexual function, fertility, or pregnancy
Staging of Wilson’s Disease
Wilson’s disease is staged using the following criteria: (This represents disease progression rather than a universally accepted clinical staging system)
- Stage 1: Initial accumulation of copper in the liver, general symptoms.
- Stage 2: Acute redistribution of copper within the liver, followed by release into the systemic circulation.
- Stage 3: Chronic accumulation of copper in extrahepatic tissues, e.g., the brain and eyes.
- Stage 4: Use of chelation therapy to restore copper balance.
Diagnosis of Wilson’s Disease
Clinicians can easily diagnose WD using blood and urine tests, eye examination, liver biopsy, genetic testing, and MRI. They use these tests together to confirm the diagnosis and assess the severity of the disease.
- Blood tests may include ceruloplasmin and serum copper. Ceruloplasmin binds copper in the blood, and gene mutations in WD prevent copper from binding to it, resulting in low levels. However, ceruloplasmin levels may be normal in some cases and should not be used alone to diagnose WD. Serum copper checks the levels of copper in the blood. These levels of unbound copper in the serum are raised in WD. Liver function tests (LFTs) are carried out to assess the functionality of the liver.
- A 24-hour urinary copper test is conducted. Your physician will ask you to collect your urine for 24 hours and test it for copper.
- Eye exam for Kayser-Fleischer rings is done via a slit-lamp.
- A sample of the liver can be taken to test for copper concentration in it. Liver biopsy measures hepatic copper concentration and assesses for damage (cirrhosis, fibrosis).
- Genetic testing to analyse the mutation of the ATP7B gene, as well as other genes linked to copper accumulation in the liver, may be performed. It confirms the diagnosis and helps with family screening.
- MRI of the brain shows a “the face of the giant panda” sign. It shows copper accumulation and damage to the midbrain.
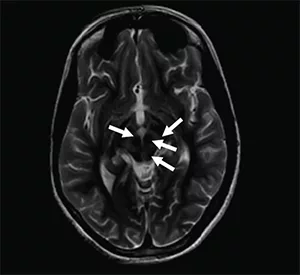
MRI brain showing the “face of the giant panda” sign in Wilson’s disease. Image Credits: Chakraborty S, Mondal S, Sinha D, Nag A.Face of the giant panda sign in Wilson’s disease.Wikimedia Commons. Licensed underCC BY 4.0.
Differential Diagnosis
Wilson’s disease’s symptoms show up as neurological or hepatic (liver) symptoms; hence, it overlaps with psychological and liver diseases. Psychological conditions that overlap with WD would include Parkinson’s syndrome, multiple sclerosis, essential tremors, etc. Liver disease includes hepatitis,Nonalcoholic Fatty Acid Liver Disease (NAFLD), liver failure, etc. Clinicians will need to exclude these diseases, and they will diagnose WD based on the above-mentioned tests.
Treatment of Wilson’s Disease
Since WD is a genetic disease, its treatment is lifelong.
Pharmacological Therapies: Chelating agents such as D-penicillamine (Cuprimine, Depen) bind to excess copper in the system and remove it via urinary excretion. Penicillamine may initially worsen neurological symptoms in some patients. If the patient does not tolerate penicillamine, Trientine (Syprine, Cuvrior) is the second-line chelating agent. Oral zinc is prescribed to prevent copper from being absorbed into the system. Zinc is also commonly used for maintenance therapy and in presymptomatic patients. Once copper reduces in the system, these agents are used in low doses for life to prevent copper buildup.
Avoid high-copper foods. These include beef livers, shellfish (oysters, crabs), chocolate, potatoes, nuts, and mushrooms.
In cases of liver failure, liver transplantation is considered.
Adjunct therapies for symptomatic treatment are also helpful. These include physiotherapies for neurological symptoms, antiepileptics in case of seizures, and neuroleptics to treat psychiatric symptoms.
Is Wilson’s Disease Fatal?
The prognosis of WD depends entirely on the time of diagnosis. If it is left untreated, copper buildups in the brain, liver, and body can lead to damage and ultimately, death. Untreated Wilson’s disease is progressive and can be fatal, often within a few decades after symptom onset. However, if detected early and treated promptly to reduce blood levels of copper, you can live a healthy and normal life. That being said, you will need lifelong medicines (chelating agents) to prevent buildups of copper in the future.
Conclusion
Wilson’s disease is a rare yet potentially life-threatening genetic disorder characterized by impaired copper metabolism due to mutations in the ATP7B gene. The resulting accumulation of copper in vital organs, particularly the liver, brain, and eyes, leads to a wide spectrum of hepatic, neurological, and psychiatric manifestations. Despite its complex presentation and overlap with other conditions, Wilson’s disease is one of the few inherited metabolic disorders that is highly treatable. Early recognition, supported by biochemical tests, imaging, and genetic analysis, is essential for preventing irreversible organ damage. With timely initiation of chelation therapy and appropriate lifestyle modifications, patients can achieve good long-term outcomes and maintain a near-normal quality of life. However, lifelong adherence to treatment and regular monitoring remain critical to prevent relapse and disease progression.
References
[1] Teschke R, Eickhoff A. Wilson Disease: Copper-Mediated Cuproptosis, Iron-Related Ferroptosis, and Clinical Highlights, with Comprehensive and Critical Analysis Update. International Journal of Molecular Sciences [Internet]. 2024 Jan 1;25(9):4753.
[2] Sandahl TD, Laursen TL, Munk DE, Vilstrup H, Weiss KH, Ott P. The Prevalence of Wilson’s Disease: An Update. Hepatology. 2020 Jan 31;71(2):722–32.
[3] Liu J, Luan J, Zhou X, Cui Y, Han J. Epidemiology, diagnosis, and treatment of Wilson’s disease. Intractable & Rare Diseases Research [Internet]. 2017 Nov 1;6(4):249–55.
[4] Pandey N, John S. Kayser-Fleischer Ring [Internet]. PubMed. Treasure Island (FL): StatPearls Publishing; 2020.
[5] Mayo Clinic. Wilson’s disease – Symptoms and causes [Internet]. Mayo Clinic. 2018
[6] Jopowicz A, Tarnacka B. Neurological Wilson’s Disease Signs—Hepatic Encephalopathy or Copper Toxicosis? Diagnostics. 2023 Feb 27;13(5):893.
[7] Ortiz JF, Morillo Cox Á, Tambo W, Eskander N, Wirth M, Valdez M, et al. Neurological Manifestations of Wilson’s Disease: Pathophysiology and Localization of Each Component. Cureus [Internet]. 2020 Nov 16;12(11).
[8] Chaudhry HS, Anilkumar AC. Wilson Disease [Internet]. Nih.gov. StatPearls Publishing; 2019.